


Abstract
Gene therapy as a potential cure for sickle cell disease (SCD) has long been pursued, given that this hemoglobin (Hb) disorder results from a single point mutation. Advances in genomic sequencing have increased the understanding of Hb regulation, and discoveries of molecular tools for genome modification of hematopoietic stem cells have made gene therapy for SCD possible. Gene-addition strategies using gene transfer vectors have been optimized over the past few decades to increase expression of normal or antisickling globins as strategies to ameliorate SCD. Many hurdles had to be addressed before clinical translation, including collecting sufficient stem cells for gene modification, increasing expression of transferred genes to a therapeutic level, and conditioning patients in a safe manner that enabled adequate engraftment of gene-modified cells. The discovery of genome editors that make precise modifications has further advanced the safety and efficacy of gene therapy, and a rapid movement to clinical trial has undoubtedly been supported by lessons learned from optimizing gene-addition strategies. Current gene therapies being tested in clinical trial require significant infrastructure and expertise, given that cells must be harvested from and chemotherapy administered to patients who often have significant organ dysfunction and that gene-modification takes place ex vivo in specialized facilities. For these therapies to realize their full potential, they would have to be portable, safe, and efficient, to make an in vivo–based approach attractive. In addition, adequate resources for SCD screening and access to standardized care are critically important for gene therapy to be a viable treatment option for SCD.
Introduction
Most therapies for sickle cell disease (SCD) are symptom focused, preventative, or disease modifying.1-4 Allogeneic blood and marrow transplantation (BMT) has been known to cure SCD but is limited by the donor pool.5-17 Gene therapy is an attractive treatment for SCD, given that the disorder results from a monogenic point mutation.18 Gene therapy is autologous and thereby avoids the inherent risks of graft rejection and graft-versus-host disease that accompany allogeneic BMT. Two general approaches to gene therapy are currently applied to SCD: (1) gene addition, where genes are transferred into the genome via a vector system, and (2) gene editing, where permanent genomic changes are made that involve removal or replacement of DNA sequences. The goal of these approaches to date has been to minimize the effect of the βS mutation by gene modifications that allow production of normal hemoglobin (Hb) or of Hb with antisickling properties. Gene modifications have specifically targeted hematopoietic stem cells (HSCs) because of their potential to provide a lifelong supply of nonsickle red blood cells (RBCs).
Therapeutic strategies
β-Globin–based strategies
One strategy for addressing sickle Hb (HbS) involves addition of β-globin genes to result in normal formation of adult Hb (HbA; Figure 1A). However, gene addition leaves the βS mutation intact; therefore, HbS would still be present in RBCs and could potentially cause SCD manifestations. This problem is also a concern with gene-editing approaches, because it is unlikely that all of the βS genes can be replaced. Addition of β-globin gene variants that also have antisickling effects has thus become favored by some groups over normal β-globin. Another advantage of such variants is the ability to track expression of the added gene, because the resulting Hb peaks can be differentiated from normal Hb by mass spectrometry. The 2 variants that are being evaluated in clinical trials are βA-T87Q, which has a single mutation conferring most of the antisickling effect of γ-globin (threonine at codon 87 replaced by glutamine) and βAS3, which has 3 mutations (antisickling T87Q and E22A and the G16D substitution that increases affinity for α-globin subunits).19,20 Other variants have been proposed and potentially have benefits in addition to antisickling, such as resistance to oxidative stress.21

Gene therapy strategies for SCD. (A) Antisickling globin expression as a gene-therapy strategy to prevent RBC sickling. (B) Gene-addition strategy to deliver antisickling genes. (C) Gene-editing approaches to induce HbF by MHEJ and gene repair by HDR.
Gene therapy strategies for SCD. (A) Antisickling globin expression as a gene-therapy strategy to prevent RBC sickling. (B) Gene-addition strategy to deliver antisickling genes. (C) Gene-editing approaches to induce HbF by MHEJ and gene repair by HDR.
γ-Globin–based strategies
γ-Globin is naturally expressed during the third trimester of fetal development and pairs with α-globin subunits to form fetal Hb (HbF). HbF continues to be expressed during early childhood and typically wanes in the first year of life.22 BCL11A serves as a switch that stops γ-globin expression naturally during the first year of life, facilitating the transition to HbA expression.23 After this period, HbF levels are typically <1% of the total Hb in RBCs. There are patients with hereditary persistence of fetal Hb (HPFH) who have mutations that lead to persistently elevated levels of 10% to 40% HbF. HPFH does not have clinical manifestations, and in those who have both HPFH and sickle cell mutations, the SCD phenotype is mild. The lack of severe disease phenotype in HPFH with SCD is believed to be related to a dilution of the HbS per RBC with HbF as well as the antipolymerizing effects that HbF has on HbS. Thus, increasing HbF levels by gene addition of γ-globin genes is a potential therapeutic approach to improve clinical manifestations of SCD (Figure 1A). Alternatively, reversing the repression of γ-globin expression by BCL11A can also increase HbF levels. Repression of BCL11A can be achieved by the addition of genes encoding short hairpin RNAs (shRNAs) with erythroid-specific promoters that result in erythroid-specific expression of shRNAs targeting BCL11A.24.25 Similarly, gene edits in the enhancer and targets of BCL11A can disrupt its effects.26,27 Thus, γ-globin continues to be produced and HbF can be formed, ameliorating the negative effects of HbS.
Gene addition
Gene-addition strategies have been investigated since the 1970s. Initially, wild-type viruses were used, after the discovery that viruses can transfer genetic information.28 With the advent of methods to transfer specific genes without the risk of viral replication, more monogenic disorders such as SCD have been evaluated for potential gene therapy (Figure 1B). In the case of SCD, HSCs have primarily been gene modified ex vivo and reinfused to avoid off-target genotoxicity in other organs that could occur with systemic delivery of gene-modifying viral vectors (Figure 2A).
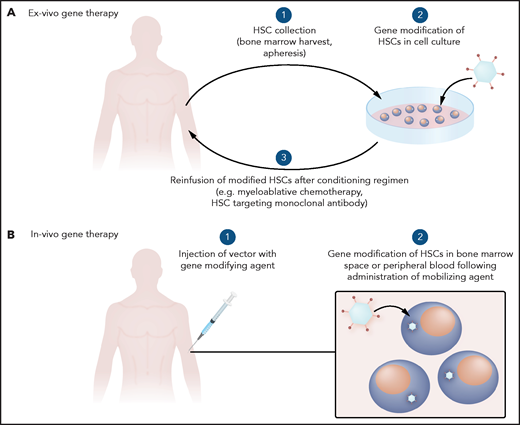
General schema for gene therapy. (A) Ex vivo therapy involves collection of HSCs to be gene modified and reinfused after a conditioning regimen. (B) In vivo gene therapy involves systemic delivery of a gene-modifying agent with trophism for HSCs.
General schema for gene therapy. (A) Ex vivo therapy involves collection of HSCs to be gene modified and reinfused after a conditioning regimen. (B) In vivo gene therapy involves systemic delivery of a gene-modifying agent with trophism for HSCs.
Gammaretroviral vectors
Retroviral vectors are based on viruses that have the ability to reverse transcribe their RNA as part of their infectious cycle and integrate genetic material into the infected cell’s genome.29 One group of retroviral vectors, gammaretroviral vectors, were used in early gene therapy trials of treatments for Wiskott-Aldrich syndrome and severe combined immunodeficiency and, in some cases, led to malignancy.30-32 Gammaretroviruses integrate near gene regulatory regions or oncogenes and have very strong promoters that contribute to their oncogenic potential. The method used for adding the gene of interest (the “transgene”) to the target cells has thus evolved in recent times to improve safety and efficacy.
Lentiviral vectors
The lentiviral vector system is the delivery method currently at the forefront of gene-addition strategies for SCD. Lentiviral vectors are retroviral vectors based on HIV; however, the genes of the native virus used to manufacture the lentiviral vector have been separated into individual plasmids.33 Transient expression of the genes on the viral plasmids generates a vector that is packaged to contain only the necessary materials to transfer the gene of interest. The viral vector therefore lacks the genes necessary for viral replication. In addition, a deletion in the 3' long terminal repeat makes the vector self-inactivating.34,35 Lentiviral vectors demonstrate improved safety compared with other retroviral vectors because they naturally integrate into actively transcribed regions of the genome, rather than near gene regulatory regions or oncogenes. Other optimizations to limit oncogenic potential include the use of native internal promoters to attain normal levels of transgene expression and the inclusion of insulator elements to prevent promoter effects of genes downstream of the transgene.36
Higher copy numbers of transgene per HSC have been associated with higher efficacy, thus prompting improvements in the manufacturing process to achieve a product with high therapeutic potential (ie, a large number of HSCs with high expression of the transgene).37 Additives such as poloxamer to improve transduction, forward orientation of transgene expression cassettes, smaller locus control regions, and pseudotyping of the vector envelope to improve cell entry are examples of methods being explored to improve the efficacy of lentiviral vector–based gene-addition strategies for SCD. 38-41
Other viral vectors
Other viral vectors are being explored to improve the efficacy and potential delivery method of the transgene. For example, adenoviral vectors such as Ad5/35++ and foamy viral vectors can be engineered to integrate genes of interest with trophism for HSCs.42,43 Although still in early development, these vectors may allow for targeting of HSCs via direct systemic delivery into the bloodstream rather than by ex vivo gene modification followed by chemotherapy conditioning and reinfusion of gene-modified HSCs. Animal data support this approach in combination with HSC mobilization techniques.44 Hurdles to clinical translation that remain with systemic delivery primarily involve the immune response and concerns about off-target effects.
Clinical trials
To date, clinical trials of gene addition have focused primarily on ex vivo gene modification, primarily because of improved feasibility, safety, and efficacy compared with a yet-to-be-developed in vivo approach (Table 1). Patients undergo HSC harvest to provide the cells for gene modification, which is performed primarily by bone marrow harvest in patients under general anesthesia. Patients with SCD need special preparation with hydration and RBC transfusion to safely tolerate the harvest procedure. Complications can occur and the cell yield may be limited by adherence of the sickle RBCs to the HSCs, making separation difficult.45 More recently, apheresis collections after HSC mobilization have been performed. This technique avoids lengthy harvests in patients under general anesthesia, although some anesthesia is still needed for central venous catheter placement to facilitate apheresis in pediatric patients. Patients often require multiple collection cycles to obtain sufficient cells for gene modification and to backup unmodified cells in case of graft failure or dysfunction of the gene-modified cells after infusion. The HSC mobilizing agent plerixafor has been used with relatively good safety in contrast to the more commonly used mobilizing agent granulocyte colony-stimulating factor, which is contradicted in SCD, as it can cause life-threatening sickling events.46.47 After collection, HSCs are gene modified in cell culture and then infused into patients who have received a conditioning regimen that makes space for the gene-modified cells in the marrow. Myeloablative chemotherapies used for allogeneic BMT are used in the conditioning regimen, given their track record in BMT for SCD (eg, busulfan and melphalan). Before the conditioning regimen, patients undergo organ function evaluation to determine whether they can tolerate the high-dose chemotherapy. Patients must also be counseled on the short- and long-term risks of these agents, including carcinogenic effects and infertility.
Ongoing gene-addition trials
| Clinical trial | Clinicaltrials.gov registry no. | Phase | Mechanism | Vector | Cell source | Conditioning regimen | Status |
|---|---|---|---|---|---|---|---|
| HGB-206 | NCT02140554 | 1/2 | βA-T87Q addition | Lentiviral | Bone marrow; plerixafor mobilized | Busulfan | Active, not recruiting |
| HGB-210 | NCT04293185 | 3 | βA-T87Q addition | Lentiviral | Bone marrow; plerixafor mobilized | Busulfan | Recruiting |
| ARU-1801 | NCT02186418 | 1/2 | γ Addition | Lentiviral | Bone marrow; plerixafor mobilized | Melphalan | Recruiting |
| Gene transfer for sickle cell disease | NCT03282656 | 1 | γ Induction via addition of shRNA silencing BCL11A | Lentiviral | Plerixafor mobilized | Busulfan | Recruiting |
| DREPAGLOBE | NCT03964792 | 1/2 | βAS3 Addition | Lentiviral | Bone marrow; plerixafor mobilized | — | Not yet recruiting |
| Stem cell gene therapy for sickle cell disease | NCT02247843 | 1/2 | βAS3 Addition | Lentiviral | Plerixafor mobilized | Busulfan | Recruiting |
| CSL200 gene therapy in adults with severe sickle cell disease | NCT04091737 | 1 | γG16D Addition | Lentiviral | Plerixafor mobilized | Melphalan | Active, not recruiting |
| Clinical trial | Clinicaltrials.gov registry no. | Phase | Mechanism | Vector | Cell source | Conditioning regimen | Status |
|---|---|---|---|---|---|---|---|
| HGB-206 | NCT02140554 | 1/2 | βA-T87Q addition | Lentiviral | Bone marrow; plerixafor mobilized | Busulfan | Active, not recruiting |
| HGB-210 | NCT04293185 | 3 | βA-T87Q addition | Lentiviral | Bone marrow; plerixafor mobilized | Busulfan | Recruiting |
| ARU-1801 | NCT02186418 | 1/2 | γ Addition | Lentiviral | Bone marrow; plerixafor mobilized | Melphalan | Recruiting |
| Gene transfer for sickle cell disease | NCT03282656 | 1 | γ Induction via addition of shRNA silencing BCL11A | Lentiviral | Plerixafor mobilized | Busulfan | Recruiting |
| DREPAGLOBE | NCT03964792 | 1/2 | βAS3 Addition | Lentiviral | Bone marrow; plerixafor mobilized | — | Not yet recruiting |
| Stem cell gene therapy for sickle cell disease | NCT02247843 | 1/2 | βAS3 Addition | Lentiviral | Plerixafor mobilized | Busulfan | Recruiting |
| CSL200 gene therapy in adults with severe sickle cell disease | NCT04091737 | 1 | γG16D Addition | Lentiviral | Plerixafor mobilized | Melphalan | Active, not recruiting |
β-Globin gene transfer
Trials testing lentiviral transfer of βA-T87Q to HSCs ex vivo are now underway. A myeloablative busulfan conditioning regimen is administered before infusion of gene-modified HSCs. One published proof-of-principle case from clinical trial HGB-205 (www.clinicaltrials.gov, #NCT02151526) involved a 13-year-old patient in France who had recurrent pain crises, acute chest syndrome, and osteonecrosis treated with chronic RBC transfusions before receiving gene therapy.37 He was reportedly transfusion free and without acute sickle symptoms at 15 months after gene therapy. His total Hb level had stabilized at 11.8 g/dL at 15 months, and the transgenic HbAT87Q comprised 48% of his total Hb, with 49% HbS. The average copy number per cell measured in the granulocyte fraction was ∼2 copies per diploid genome and was stable between 12 and 15 months without evidence of clonal outgrowth. Outcome data on 3 cohorts of the US-based HGB-206 Study (#NCT02140554) have also been presented.48,49 In the first cohort, 6 of 7 patients had vector copy numbers <1 in the gene-modified product, and although the transgenic Hb level rose in the patients’ blood over time, the average level was less than 1 g/dL at 36 months. With subsequent cohorts, the manufacture and cell-collection processes were improved and included a change from bone marrow harvest to apheresis to optimize HSC yield. After this optimization, 25 patients were treated with an increased vector copy number (median, 3.8; range, 2.3-5.7) in the gene-modified product. In 16 patients with ≥6 months’ follow-up, the Hb ranged from 9.6 to 016.2 g/dL, transgenic Hb ranged from 2.7 to 9.4 g/dL, and the median HbS was ≤60% with HbAT87Q of ≥40%. The median annualized rate of vaso-occlusive events decreased from 4 to 0 after infusion. After gene therapy, 1 patient with a history of pulmonary hypertension died of sudden-onset cardiac arrest that was attributed to complications from longstanding SCD and not to gene therapy. There has been no evidence of clonality to date in patients with gene-engrafted cells in the new cohort. In the first cohort where patients had minimal gene-modified cells remaining, a patient developed myelodysplasia and another developed acute myeloid leukemia, and further studies were performed to determine which component of gene therapy contributed to malignant transformation and to guide refinements in the therapeutic approach. Based on an absence of transgene in the myelodysplastic cells and in accordance with recent recognition of myelodysplasia as a potential risk in adults who have graft failure after nonmyeloablative allogeneic BMT for SCD, myelodysplasia has been attributed to the gene therapy conditioning regimen (busulfan) rather than to the gene therapy vector,50,51 prompting efforts to optimize conditioning as a means of improving safety. In the patient with leukemia, vector insertion was seen in a gene not associated with malignancy suggesting the leukemia was not vector-mediated and further investigation is ongoing.52
γ-Globin gene transfer
The basis of the ongoing phase 1/2 pilot study of ARU-1801 (#NCT02186418) is to develop a gene therapy approach that would achieve sufficient engraftment of HSCs that have undergone addition of lentiviral vector γ-globin, while using a less intense conditioning regimen of melphalan (140 mg/m2 IV, once) rather than busulfan.53,54 The advantages of this approach include the portability of melphalan and its single dose based on body surface area. Busulfan requires multiple doses and blood draws for drug level monitoring and real-time dose adjustment. In this trial, HSCs have been collected by marrow harvest and/or apheresis. The first 2 patients had a vector copy number of 0.2 to 0.4 detected in bone marrow and peripheral blood at 15 and 12 months, respectively. The first patient had a vector-derived HbF of 20% and a total Hb level of 10.6 g/dL at 1 year, which was a substantial improvement over the baseline Hb of 7.5 to 8.5 g/dL. A similar HbF pattern was seen in the second patient but at a lower trajectory. Both patients had a >90% reduction in acute sickling episodes after treatment. Outcome data on an additional 2 patients are pending. Although the increase in HbF in this trial to date has been modest, it demonstrates that disease palliation rather than cure may be acceptable. In addition, the conditioning regimen was well tolerated (as expected), and there were no concerns about clonality or abnormal hematopoiesis.
BCL11A silencing
The goal of this trial (#NCT03282656) is HbF induction by silencing BCL11A in erythroid cells.55 This induction is achieved by ex vivo HSC lentiviral transduction to transfer an shRNA transgene driven by an erythroid-specific promoter. Cells were collected for gene modification by plerixafor mobilization and are infused after a conditioning regimen of myeloablative busulfan. In the pilot phase of the study, 3 adults, 2 adolescents, and 1 pediatric patient were treated. In 1 adult patient who had moya central nervous system disease, long-term RBC transfusion therapy was resumed 3 months after infusion because of an HbS level of >40%. Before the treatment, the HbF level was 40.7%. The other patients in the pilot study had HbF levels of 22.7, 31.9, 38.8, 29 and 41.3%, at 24, 21, 12, 12 and 6 months of follow-up, respectively. There were no vaso-occlusive events after gene therapy except priapism in 1 patient; a larger scale trial is planned.
Gene editing
Editing the genome remains conceptually very appealing for monogenic disorders such as SCD, because repairing a mutation would lead to a cure. A variety of molecular tools have been developed in recent years to facilitate genome editing, and these tools continue to advance rapidly. The most used editors (meganucleases, zinc finger nucleases, TALENS, and CRISPR-Cas) involve making double-strand breaks at a specified location in the genome that result in natural repair mechanisms.56 Delivery mechanisms for gene editors have included viral vectors, electroporation, and nanoparticles delivered into ex vivo collected HSCs.26,57-59
Nonhomologous end joining
The predominant and error-prone version of double-stranded DNA repair is nonhomologous end joining (NHEJ). With this approach, repair occurs mostly at the cost of loss of gene function in the region where the breaks happen. Double-strand breaks combined with the NHEJ that disrupts gene function can be used for reinduction of HbF by removing the silencing that occurs developmentally in the transition to HbA. Examples of gene edits to induce HbF in this way include disruption of (1) erythroid-specific enhancer regions of BCL11A, (2) HBG 1 and 2 promoter regions, and (3) KLF-126.27,60,61 (Figure 1C).
Homology-directed repair
The less common mechanism of repair, called homology-directed repair (HDR), makes use of a template to potentially restore the damaged gene’s ability to encode a normal protein. Gene-editing approaches that involve HDR require delivery of nucleic acid sequences that can be used as repair templates that introduce a desired DNA sequence where the double-strand break has been made. In this way, a genomic region harboring a mutation can be repaired by replacement with a normal one. For example, in SCD, β-globin edits are made to excise the sickle mutation and the donor templates can be either normal β-globin sequences or variants with antisickling properties57,62 (Figure 1C).
HDR and NHEJ can occur simultaneously, resulting in gene correction in some cases and gene disruption in others. A reduction in β-globin genes caused by NHEJ can thus lead to low Hb formation and a clinical concern of β-thalassemia. Although genome editors have fairly precise targets for DNA break points, repair efficiency with HDR is low, and off-target breaks remain a concern. Complex on-target DNA repair events are also possible, although their frequency and significance after HSC editing remains uncertain.63
Base editing
A newer group of genome editors called base editors are tools that can make individual nucleotide edits on a single strand of DNA and thus may be safer than double-strand breaks.64 Currently, there are 2 types of base edits than can be made: converting C to T or A to G. Because the SCD mutation is an A-to-T mutation, it cannot be directly repaired with currently available base editors. Thus, work in SCD has focused on making edits that would favor HbF induction, although one could instead edit to produce a variant β-globin.65
Off-target effects
The detection of off-target effects is uniquely challenging and becomes more difficult the smaller the edits are, such as with base edits. DNA sequencing technology has improved significantly and can detect sequence changes at the genome level; however, there remains significant debate as to the best assays to measure off-target effects related to sensitivity and specificity concerns.66 Further, off-target effects detected by in vitro assays may not be representative of in vivo models, and human studies are needed to determine the clinical impact of potential off-target edits.
Clinical trials
There are fewer clinical data available on gene editing compared with gene addition, as gene editing is a relatively recent discovery, with limited clinical translational research to date (Table 2). In general, the trials that are being developed for gene editing mirror those for gene addition, in that HSCs are collected from the patient for gene modification ex vivo and are reinfused after myeloablative chemotherapy. In addition, gene-editing strategies aimed at disrupting genes are more efficient than strategies aimed at repairing genes, thus most trials that are initiated or planned involve reactivation of fetal Hb by disrupting a repressor.
Ongoing gene-editing trials
| Clinical trial | Clinicaltrials.gov registry no. | Phase | Mechanism | Gene editor | Cell source | Conditioning regimen | Status |
|---|---|---|---|---|---|---|---|
| CLIMB-SCD-121 | NCT03745287 | 1/2 | Erythroid lineage-specific enhancer of the BCL11A disruption | CRISPR-Cas9 | Plerixafor mobilized | Busulfan | Recruiting |
| PRECIZN-1 | NCT03653247 | 1/2 | BCL11A locus targeting | Zinc finger nuclease | Plerixafor mobilized | Busulfan | Recruiting |
| Clinical trial | Clinicaltrials.gov registry no. | Phase | Mechanism | Gene editor | Cell source | Conditioning regimen | Status |
|---|---|---|---|---|---|---|---|
| CLIMB-SCD-121 | NCT03745287 | 1/2 | Erythroid lineage-specific enhancer of the BCL11A disruption | CRISPR-Cas9 | Plerixafor mobilized | Busulfan | Recruiting |
| PRECIZN-1 | NCT03653247 | 1/2 | BCL11A locus targeting | Zinc finger nuclease | Plerixafor mobilized | Busulfan | Recruiting |
In the CLIMB-SCD-121 trial, HSCs are collected by apheresis, edited by a CRISPR-Cas9 system targeted to disrupt an erythroid enhancer region of BCL11A, and lead to HbF induction. Gene-modified cells are infused after busulfan myeloablation. To date, results from a single patient with an annualized history of 7 vaso-occlusive events were reported. At 15 months after treatment, the patient was reported to be free of vaso-occlusive events and the HbF level was 43.2%, HbS level was 52.3%, and the total Hb was 12 g/dL, compared with 9.1%, 74.1%, and 7.2 g/dL, respectively, at baseline. There have been no reports of safety issues related to the gene-modified product, and the study is progressing with a second patient treated and the cells engrafted.67
The PRECIZN-1 trial is another currently open study that is an investigation of zinc finger nuclease disruption of the erythroid enhancer of BCL11A to induce fetal Hb.68 The clinical results of this trial are still pending.
Considerations for the curative potential for gene therapy
After allogeneic BMT, symptoms of SCD resolve if 20% to 25% of HSCs in the bone marrow space are of donor origin.69,70 This resolution occurs, even though most of the HSCs are of recipient origin harboring homozygous βS mutations and donor HSCs carrying heterozygous βS mutation (sickle trait donors). The RBC progeny of the donor HSCs have substantially longer half-lives than that of the SCD recipient HSCs, resulting in only normal RBCs in the peripheral blood and resolution of SCD. Thus, for gene therapy to be curative, at least 20% to 25% of HSCs in the bone marrow must be modified in a manner that leads to functionally normal RBCs. To achieve this, nonsickle Hb levels in each RBC derived from gene-modified HSCs would have to be sufficiently high to prevent sickling. A heterocellular distribution of nonsickle Hb, however, would theoretically lead to disease amelioration rather than cure. In gene-addition strategies, curative intent thus relies on achieving a sufficient vector copy number in at least 20% to 25% of HSCs that allows for a level of therapeutic Hb expression that prevents sickling and normalizes the red blood cell lifespan. In gene editing for HbF induction, curative intent relies on achieving sufficient edits again in 20% to 25% of HSCs that result in essentially all RBCs in the peripheral circulation having sufficiently high and homogenous HbF expression that prevents sickling and normalizes the RBC lifespan. This lofty goal differs from the disease amelioration effect of hydroxyurea in SCD where HbF expression on average in RBCs can reach 20% to 30% but with cell-to-cell HbF expression being quite variable. The RBC progeny of gene-modified HSCs using optimized lentiviral transfer of βA-T87Q demonstrates a reciprocal decrease in HbS per cell, and preliminary studies in patients have demonstrated HbS expression less than that in RBCs from persons with sickle trait.71-73 These data provide support for such an approach being potentially curative. However, long-term clinical efficacy of gene therapy to cure SCD is still in question, given that data are available only from early-phase clinical trials.
Considerations for broad applications of gene therapy for SCD
Eligibility
Traditionally, patients with SCD have had to meet disease burden criteria to qualify for curative allogeneic BMT, although these criteria were set by expert consensus rather than by evidence, and this area remains in need of research. Gene therapies are being offered solely in clinical trials and the use inclusion and exclusion criteria have been based on allogeneic BMT. However, patients with significant central nervous system complications are generally excluded, given that it is unclear whether gene therapy is sufficiently efficacious for these patients. In addition, patients with significant organ dysfunction are excluded, given the need for myeloablative chemotherapy in most gene therapy trials. Use of gene therapies will be limited while they are available only through clinical trials and are approved only for narrow indications. Further research and longer follow-up are needed to develop inclusion criteria that balance the significant risks with the yet-to-be-determined benefits of gene therapy for individual patients.
Safety
HSC collection is not a benign procedure in SCD, given the associated risk of severe vaso-occlusive events. However, improvements in HSC mobilization, including newer and potentially more efficacious mobilizing agents, will reduce the number of cycles of apheresis that patients have to undergo. Conditioning chemotherapy regimens that are currently used to make space in marrow for the gene-modified HSCs may deter or limit access for older patients with organ dysfunction or for those patients concerned about the relatively high risk of infertility. There is also the risk of malignancy and establishing the contributing component of gene therapy involved (vector vs conditioning regimen) is critical for determining how to improve the approaches to lessen these risks. For example, if malignancy was vector derived, then one would endeavor to optimize vector safety with additional elements in the vector design. If malignancy was conditioning related, then one would ensure complete myeloablation to avoid residual HSCs with genetic lesions from the chemotherapy undergoing malignant transformation and/or focus on antibody-based conditioning regimens (eg, anti-CD117).51,74 These important risks should be weighed in the context of gene therapy being in its infancy, with only early results from a few trials available currently. Thus, the US Food and Drug Administration has mandated 15 years of follow-up to determine these and other risks.
Feasibility
The current strategy of ex vivo gene modification combined with high-dose chemotherapy is best suited for use in centers in developed nations with expertise in hematology and transplant for SCD. This model would need to be adjusted for widespread use in the developed and developing world, especially given that most patients with SCD are in low-resource settings worldwide. Systemic delivery of a vector for in vivo gene modification (Figure 2B) may avoid the need for a conditioning regimen and the associated intense supportive care infrastructure. Although systemic delivery could make gene therapy more portable, challenges of overcoming an immune response to the gene-modification tools, while ensuring efficient targeting to the cells of interest would have to be addressed. The cost of gene therapy is likely to be decided by weighing important contributing factors, such as cost savings for the health system, cost recovery for industry partners who make large investments to get therapies approved, and affordability for patients worldwide.
Summary and conclusion
Significant advances in gene-addition strategies over the past few decades, including successful translation to clinical application, have paved the way for gene-editing approaches. The coming years are likely to see approvals for multiple gene therapies. There remains, however, a critical need to improve access to standard care (screening, medications, vaccines, and clinical infrastructure) to make gene therapy for SCD more accessible as most patients worldwide are in low-resource settings. Future research is needed to optimize efficacy, evaluate long-term safety, and improve the feasibility and portability of gene therapy. These efforts will be crucial in the development of an efficacious in vivo approach for SCD. One thing is clear: after decades of research, gene therapy is advancing as a promising therapy for SCD.
Acknowledgments
This work was supported by the American Society of Hematology and the Robert Wood Johnson Foundation (Amos Medical Faculty Development Program) (A.A.A.); and the Intramural Program of the National Institutes of Health, National Heart, Lung, and Blood Institute (grant HL006008-07) (J.F.T.).
Authorship
Contribution: A.A.A. and J.F.T. wrote the paper.
Conflict-of-interest disclosure: A.A.A. has served on the safety monitoring committee for Sangamo Therapeutics and has no financial interest in the development of gene therapies. J.F.T. declares no competing financial interests.
Correspondence: Allistair A. Abraham, Children’s National Hospital, 111 Michigan Ave, NW, Washington, DC 20010; e-mail: aabraham@childrensnational.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal