

Abstract
In chronic lymphocytic leukemia (CLL), increasing knowledge of the biology of the tumor cells has led to transformative improvements in our capacity to assess and treat patients. The dependence of tumor cells on surface immunoglobulin receptor signaling, survival pathways, and accessory cells within the microenvironment has led to a successful double-barreled attack with designer drugs. Studies have revealed that CLL should be classified based on the mutational status of the expressed IGHV sequences into 2 diseases, either unmutated (U) or mutated (M) CLL, each with a distinctive cellular origin, biology, epigenetics/genetics, and clinical behavior. The origin of U-CLL lies among the natural antibody repertoire, and dominance of IGHV1-69 reveals a superantigenic driver. In both U-CLL and M-CLL, a calibrated stimulation of tumor cells by self-antigens apparently generates a dynamic reiterative cycle as cells, protected from apoptosis, transit between blood and tissue sites. But there are differences in outcome, with the balance between proliferation and anergy favoring anergy in M-CLL. Responses are modulated by an array of microenvironmental interactions. Availability of T-cell help is a likely determinant of cell fate, the dependency on which varies between U-CLL and M-CLL, reflecting the different cells of origin, and affecting clinical behavior. Despite such advances, cell-escape strategies, Richter transformation, and immunosuppression remain as challenges, which only may be met by continued research into the biology of CLL.
Introduction
During the past 35 years, progress in understanding the biology of chronic lymphocytic leukemia (CLL) has transformed our management and treatment of patients with this disease. Although tumor-associated genetic lesions are of clear importance, much of the progress has been achieved through a better understanding of the cellular biology. The fact that CLL is a relatively indolent tumor, readily obtained from the blood or marrow, has allowed investigation into early tumor behavior before the perturbation of therapy. It also has enabled comparison with the likely normal counterparts, CD5+ B cells.1
The picture that has emerged illustrates a fundamental principle: while initial genetic changes provide developing tumor cells with a backdrop of protection against cell death, the forces underpinning the growth and survival of the neoplastic cells are mainly shared with that of their normal B-cell counterparts. The influence that the B cell of origin has on tumor behavior is striking and persistent, operating until later stages, when proliferation allows accumulation of other, often varied genomic changes that provide a selective survival or growth advantage to subclones of the tumor population.2 The cells giving rise to CLL originate from 2 different stages of B-cell differentiation distinguished by the absence or presence of somatic mutations in the rearranged immunoglobulin variable region (IGV) genes of the B-cell receptor (BCR). This finding revealed that the B cell of origin has a strong influence on the subsequent clinical behavior of the tumor.3,4
Therapy, when required, can now be calibrated to target 2 main vulnerabilities, BCR signaling and anti-apoptotic pathways, with major successes already evident.5,6 This Perspective offers a view of the evolution of CLL and integrates the biology with the clinical effects of precise targeting of the pathways essential for growth/survival of tumor cells.
Overcoming bottlenecks in tumor development
Tumor cells in general are opportunistic and variable in exploiting mutational changes and the surrounding microenvironment. A primary bottleneck for B-cell tumorigenesis, in general, is protection against death, a fate that awaits unselected normal B cells exposed to antigen. Indeed, it is estimated that we make >100 million B cells each day, with estimated daily proliferation rates ranging from 0.46% to 2.66% for naive and memory-type B cells.7 Most of these newly formed B cells will incur an unceremonious death unless they activate survival pathways induced through the BCR and other surface receptors. Activation of such pathways is also important for the survival of neoplastic CLL B cells, providing support for their clonal expansion but also revealing an Achilles heel that can be targeted by newly developed therapies.
Upregulation of BCL2 plays an important role in mitigating the normal tendency of B cells to undergo apoptosis. This requirement appears to be the case for many B-cell tumors, where remarkably different strategies can be engaged to maintain expression. CLL cells express high levels of BCL2, more than other B-cell malignancies, such as follicular lymphoma (FL), which commonly harbors chromosomal translocations that juxtapose the BCL2 locus on chromosome 18 with the IG heavy chain (IGH) promoter present on chromosome 14, representing the t(14;18) translocation. Although CLL cells of a few patients may harbor similar translocations,8 these are much less frequent than in FL.9 Instead, CLL cells more commonly (∼80% of cases) have loss or repressed expression of 2 non-coding microRNAs, namely miR-15a/16-1,10 mainly due to del(13)(q14). Since these microRNAs ordinarily repress the expression of BCL2,11 their loss or repression accounts in part for the very high-level expression of BCL2. However, although they generally have a higher expression of BCL2 than the neoplastic cells in FL, CLL cells have a greater sensitivity to the cytotoxic effect of BCL2 inhibitors, such as the BH3 mimetic venetoclax.12 This may be partly due to expression of high levels of BH3-only pro-apoptotic molecules, such as BCL2-interacting mediator of cell death (BIM). This sequesters BCL2 and primes CLL cells for death, causing CLL cells to balance between life or death on the razor’s edge of the BCL2:BIM ratio.13
The second bottleneck for CLL is to express a BCR “tuned” for stimulation of growth and survival. The BCR is composed of antigen-binding surface immunoglobulin (sIg) and accessory molecules required for BCR signaling upon ligation of the sIg. A functional BCR is required for the survival of normal B cells14 and for most mature B-cell malignancies, including CLL. The BCR of CLL cells appears engaged by environmental or self-antigen, leading to constitutive signaling in vivo.15-17 However, this is not a simple on-off switch; cellular outcome for B cells depends on the level of BCR signaling, as seen in B-cell acute lymphoblastic leukemia, where high levels of sIg ligation can induce cell death.18 This has also been shown for a murine B-cell lymphoma19 and normal memory B cells, which, in the absence of CD4+ cells, may be susceptible to “death by antigen” mediated via mitochondrial dysfunction and accumulation of reactive oxygen species.20 On the other hand, too little stimulation fails to provide the signal required for the metabolic demands of tumor cells. Clearly, an adequate level of engagement is required, so the ideal lies somewhere in the middle. This is reminiscent of the story of Goldilocks and the 3 bears, where only the porridge that is not too hot or too cold but is “just right” is acceptable.21 It is becoming clear that this Goldilocks principle applies to many biological systems, including T-cell responses to pathogens in the gut22 and the control of critical intracellular pathways such as phosphatidylinositol 3-kinase.23
One way of achieving this Goldilocks level for CLL cells is likely to be a reduced level of expression of sIg (∼10% of normal B cells) and expression of inhibitors of BCR stimulation such as CD5 and FcgRIIb.24 Many other factors, including ZAP-70, may also modulate signaling, and the importance of the consequently tuned BCR pathway to CLL cells is amply illustrated by the therapeutic success of targeted inhibition.
The cytogenesis of CLL
In 1996, a review of CLL by Guillaume Dighiero concluded by saying “Unfortunately, the results so far still give no clear indication from which B-cell population the CLL cells are derived.”25 This was about to change dramatically due to the ability to sequence IG DNA/complementary DNA and the mapping of human germline IGV, D (diversity), and J (joining) regions in the 1990s.26,27 The analysis of IGHV(D)J rearrangements in CLL cases revealed 2 critical features: (1) asymmetric usage of available IGHV regions, with predominance of IGHV1-69 (51p1)28 ; and (2) ∼40% of cases had unmutated genes (>98% homology to germline sequence, a cutoff selected to allow for polymorphisms, with the remainder having significant levels (<98% homology) of somatic hypermutation.29 This led to the designation of 2 subsets of CLL, unmutated (U) and mutated (M).3,4
CLL cells with unmutated IGHV were deduced to originate from a B cell that had not undergone somatic hypermutation in the IGV(D)J region, a process known to occur in the germinal center.30 On the other hand, CLL cells with mutated IGV apparently arise from a post-germinal center B cell. A minority of cases (6% to 10%) had also undergone isotype switch recombination.29
It has been suggested that the percent IGHV germline homology be considered more of a continuous variable based on outcome analyses of patients treated with fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy.31 However, the relatively few CLL cases with intermediate levels include those that express IGHV3-21 with 96% to 98% germline homology; such cases have a clinical course that is more typical of those that use an unmutated IGHV.32 They could originate from B cells that have exited early from the germinal center.33 In any case, the working definition for unmutated IGHV of ≥98% homology a reliable threshold for the majority.
The predominating IGHV1-69 cases are almost entirely in U-CLL, and in this subset, there is evidence for restriction in usage of IGHD and IGHJ, leading to the concept of “stereotypic” sequences, in some cases with selected IGV light chains, indicating a shared antigen specificity.34,35 Asymmetry of IGHV is less striking in M-CLL, with the initially suggested predominance of IGHV4-34 likely due mainly to increased usage in the normal aged repertoire.36 In contrast, there is a clear preferential usage of IGHV4-34 in the subset of CLL that has undergone isotype switching to IgG.36
While this information provided biological insight, identification of the 2 subsets electrified the clinical community as tumor progression differed dramatically, with cases of U-CLL having a more aggressive disease and a worse prognosis than patients with M-CLL.3,4 In the vast majority of cases, the IGHV mutational status of CLL cells is fixed, reflecting little or no interchange between the 2 subsets. It confirms that the parental B cell influences subsequent tumor behavior, as is the case for the wide range of tumors that develop from B cells at distinctive stages of differentiation.
Accumulation of genomic mutations will be influenced by the rate of proliferation, which is higher in U-CLL, where most of the clinically relevant changes are found.37 Although it is not resolved whether activation-induced cytidine deaminase is involved in generating such mutations, an activation-induced cytidine deaminase–associated signature is actually more evident in M-CLL.38
In the past, much effort was put into chasing the cell of origin of CLL by phenotypic similarity. It does appear that CD5+ B cells might be the source of both U-CLL and M-CLL.1 However, while expression of CD5 is associated with B1 cells and with anergy in mice, this is less clear for human B cells.39 Also, tumor cells may modulate their surface receptors as they interact with the leukemic microenvironment. One feature that has been revealing is the pattern of DNA methylation; the striking difference in profile between normal naive and memory B cells is, perhaps surprisingly, maintained in U-CLL and M-CLL.40 It appears that an epigenetic memory of the B cell of origin persists through transformation and marks the 2 subsets.41 Again a minority of cases show an intermediate signature, but these include the outlier IGHV3-21 cases. Clearly, tumor-related changes also occur in the epigenome, but the influence of the parental B cell is retained.42
Clues from IGHV1-69 predominance in U-CLL
The prevalent usage of IGHV1-69 is tumor related, as there is no increase in the aged population.43 There is also apparent selective use of certain alleles of IGHV1-69 in CLL.44 Moreover, the mean number of codons encoding the third complementarity determining region (CDR3) of the IGHV1-69 genes tends to be greater than that of the normal adult B-cell repertoire.44 This is partly due to preferential usage of IGHJ6 in ∼57% of IGHV1-69–expressing cases as compared with ∼29% in normal B cells.43-45 Any asymmetry in expression of an IGHV indicates a drive on the framework region, possibly also extending to CDR3 and IGHJ. Large multivalent superantigens can bind in this way and, for IGHV1-69, can be derived from pathogens such as Streptococcus pneumoniae, cytomegalovirus,46 hepatitis C virus,47 or self-antigens.48,49 It results in conserved sequences in the IGHV of stimulated B cells exactly as found in CLL. In fact, it was possible to detect very similar sequences in normal blood and spleen,43,50 thereby revealing the likely cell of origin.
This location and certain shared features point to B cells expressing the natural antibody repertoire as the source of U-CLL.34 In addition to IGHV1-69, this repertoire includes a range of other IGHV and conserved sequences, and several are mirrored in CLL. Natural antibodies are generally IgM with unmutated IGHV and show a broad low affinity/high avidity innate reactivity, often polyreactive, against common pathogens.51 The expressing cells are mainly CD20/CD27/CD5, with moderate levels of CD43/CD38, very similar to U-CLL.52 Production of natural antibody is rapid, providing immediate protection against infections and, importantly, considered to be T-cell independent. Intriguingly, natural antibodies often display autoreactivity, and they appear to be involved in the clearance of apoptotic cells or misfolded proteins, indicating a role in regulating inflammation and maintaining immune homeostasis.52
The crossover between infection and autoimmunity is nicely illustrated for IGHV1-69, which is preferentially used by antibodies against hepatitis C virus and a co-induced autoreactive IgM anti-IgG rheumatoid factor (RF).34,47,53 The complexes formed among viral antigens, antibodies, and RF can lead to mixed cryoglobulinemia with fixation of complement causing a purpuric rash.54 In this case, chronic infection has induced an IGHV1-69–encoded pathological autoantibody. sIgM with similar RF autoreactivity also is expressed by CLL cells,55 again supporting a link to natural antibodies.
The next question is how the B cells expressing IGHV1-69 develop into CLL. There has been much debate around the relationship of monoclonal B-cell lymphocytosis (MBL) to CLL, and there is evidence for clonal B-cell expansions in older age.56,57 However, low-count MBL tends to be of IGHV-mutated B cells and does not include significant levels of IGHV1-69,58 suggesting that these are not the precursors of U-CLL. It is more likely, although unproven, that they relate to M-CLL. In contrast, high-count MBL (B-cell count >0.5 × 109/L) can be of B cells with unmutated IGHV that include IGHV1-69 at frequencies similar to U-CLL, and these invariably progress to overt CLL.59,60
Function of the BCR in CLL
The BCR is the key receptor for all B cells, and CLL is no exception. There is evidence for activation of the BCR pathway in vivo, and targeting of the intracellular pathways has shown striking clinical success.61 The most advanced data are from ibrutinib, which is aimed at Bruton tyrosine kinase (BTK), and, although not entirely specific for the BCR, the clinical results support its importance and are impressive.
It turns out that the status of the BCR varies in CLL, with evidence of repetitive engagement by a ligand, presumably a self-antigen, in vivo. There is clear downregulation of expression of sIgM, especially in M-CLL. Recovery of expression occurs in vitro, consistent with reversible endocytosis.16 The resulting levels of sIgM on the circulating CLL are variable in vivo and lead to a rheostat of signaling capacity.62 This is analogous to normal B cells undergoing chronic antigenic stimulation, where downregulation of BCR expression is accompanied by a low level activation of intracellular pathways.63 A state of anergy is induced with increased levels of the negative regulator SHIP1 and NFAT2 and reduced responses to chemokines and Toll-like receptor ligands.39 A similar variable increase in NFAT2 is evident in anergized CLL cases, and, in parallel with the increase in sIgM, this is reversed by incubation in vitro.64
Anergy clearly exists in CLL at different levels, being more marked in M-CLL. In fact levels of expression and function of the residual sIgM in CLL can provide a useful prognostic marker.65 It could be relevant especially within M-CLL, where there appears to be a significant proportion of patients with deeply anergic tumor cells,16 who may not require treatment, but others with levels that approach those of U-CLL, who are projected to progress to treatment requirement and need to be monitored in clinic over time.65
The hypothesis that the tumor BCR is influenced by chronic exposure of antigens at tissue sites has been reinforced by studies in CLL patients receiving ibrutinib.66 By inhibiting BTK-dependent pathways, including BCR-mediated signaling/adhesion, and CXCL12-induced migration,67 ibrutinib redistributes leukemic cells from tissue to peripheral blood.68 There, the CLL cells specifically increase levels of sIgM, mimicking the recovery in vitro.16 Re-expressed sIgM retains signaling function at the level of phosphorylation of SYK upstream of BTK blockade.65 There is also the reconstitution of complex N-glycosylated structures in the constant region, another feature of disengagement from antigen.69 These phenomena are selective for sIgM and contrast with the expression of sIgD and of the majority of other receptors that gradually decline.66 The increase of sIgM levels and signaling capacity, with no change in sIgD, is again a defining feature of recovery from the anergic state preinduced by chronic antigen engagement in normal B cells.63 Among the other receptors, CD20 is one of the most downmodulated during therapy,65 possibly via inhibition of an activating SDF-1–mediated CXCR4 signal.70
It is in the peripheral blood that ibrutinib-treated CLL cells will lack the nourishing signals from (self-)antigen, consigning them to limbo, where apoptosis might be expected and is evident from induction of BIM expression.66 However apoptosis could be slowed by constitutionally abundant BCL2.66 Concomitantly, autophagocytic events are also promoted, including cell contraction and induction of the autophagosome-associated LC3B-II.65 The majority of CLL cells will eventually die, but cells with relatively higher sIgM levels may overcome BTK blockade by ibrutinib.71 A fraction also will have high CXCR4 levels.62 If they maintain some migratory capacity, CLL cells with high sIgM and CXCR4 levels may traffic to tissue, reencounter antigen, and receive BCR-induced signals able to circumvent BTK blockade and eventually drive a re-expansion of the disease. Any such escaping cells might be those to target by combined therapeutical approaches, including BH3 mimetics.6
However, the main recognized mechanism linked to disease re-expansion during ibrutinib is the accumulation of mutations in the BCR-associated kinases BTK and PLCγ2. BTK mutations affecting the cysteine residue at position 481 disrupt ibrutinib binding and are the most common lesions.72 PLCγ2 mutations are less frequent and appear to confer resistance mainly by activating pathways not involving BTK.73,74 In fact, CLL cells with strong BCR activation can circumvent catalytically inactive BTK to mediate increases in [Ca2+].75 Resistance also can involve expansion of preexisting subclones with a suite of genetic changes.74
Clearly, inhibition of BTK presents a not-insuperable block on the ability of some CLL cells to generate the Goldilocks level of signaling. Tumor cells may bypass BTK73,74 or activate alternative strategies via mutation.
Migration to tissue sites
CLL cells migrate from blood to marrow and lymph node sites, where proliferation occurs. Entry to lymph nodes is via high endothelial venules (HEVs), and CLL cells can be seen in crowded pockets following extravasation.76 Transmigration depends on l-selectin–mediated interaction with endothelial sialomucins, followed by chemokine-induced activation of integrins. In fact, upregulated CD49d/VLA4 and CXCR4 are known negative prognostic factors even within M-CLL.77,78 Sensing and response to dendritic cell–associated (self-)antigen by B cells can occur soon after extravasation and before migration along “exit ramps” formed by fibroblastic reticular cells, which traffic cells to the follicles.76 Retention of CLL cells in the lymph node may be extended because of reduced expression of sphingosine-1-phosphate receptors, particularly in U-CLL, thereby prolonging tissue-based nourishing signals.79 The ability of ibrutinib and possibly other drugs to inhibit migration and adhesion67 may contribute to the accumulation of CLL cells in the blood after treatment.
Microenvironmental influences
CLL cells in the lymphoid tissues form so-called proliferation centers15 instead of normal germinal centers. In such sites, CLL cells interact with stromal cells, nurse-like cells (lymphoma-associated macrophages), and T cells. The multiple influences of surrounding cells are shown in Figure 1.80 Signaling derived from ligation of the BCR by environmental or self-antigen can induce B-cell activation and proliferation in tissue sites, but only a small percentage of the CLL cells may undergo proliferation, with the remainder being either unstimulated or driven into anergy.81 Nonetheless, within these sites, all CLL cells can be stimulated by chemokines, integrins, cytokines, and survival factors (such as tumor necrosis factor [TNF] ligand superfamily member 13B, also known as B-cell activation factor [BAFF] or TNF ligand superfamily member 13 [APRIL]), which activate canonical NF-κB.82
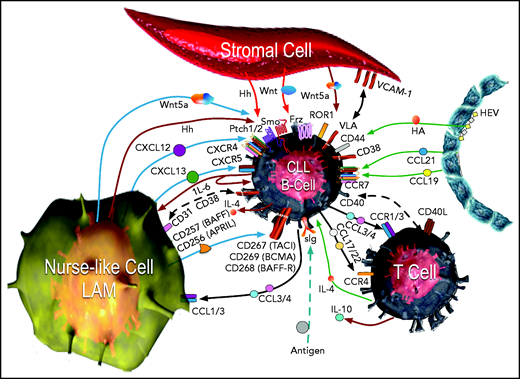
The CLL microenvironment. The main types of accessory cells in the CLL microenvironment are depicted surrounding the CLL B cell, which is located at the center.33 CLL cells enter the lymphoid tissue through HEVs (depicted on the right), which also may elaborate chemokines, such as CCL21 and CCL19, which bind CCR7 and can attract CLL cells. HEVs also produce hyaluronic acid (HA), which can activate CD44 on the CLL cell, as it attaches to HEVs via l-selectin (CD62-L) to egress from the blood (CD62-L). In the bottom left corner is a nurse-like cell (NLC), a lymphoma-associated macrophage (LAM) that secretes a number of chemokines, such as CXCL12 or CXCL13, which bind to CXCR4 or CXCR5, respectively, on the CLL cell, attracting it to the microenvironment. The leukemia cell in turn can elaborate chemokines, such as CCL3 and CCL4, which bind to CCR1 and CCR3 (CCR1/3), or CCL17 and CCL22, which bind CCR4, to recruit T cells (depicted in the bottom right) and monocytes, which are the cells that can differentiate into NLCs. Cognate interactions between CD31 and CD38 on NLCs and CLL cells, respectively, may promote CLL-cell survival. The NLC also expresses proteins of the TNF family, such as BAFF (CD257) and a proliferation inducing ligand (APRIL or CD256), which in turn activate TNF-family receptors such as transmembrane activator and calcium-modulator and cyclophilin ligand (CAML) interactor (TACI or CD267), B-cell maturation antigen (BCMA or CD269), or BAFF-receptor (BAFF-R or CD268) on the CLL cell, inducing activation of nuclear factor κ-light chain B (NF-κB). NLCs and stromal cells (depicted on top) also can produce factors that stimulate the CLL cell, such as various Wingless-related integration site (Wnt) factors, with interact with Frizzled (Frz) receptors, or noncanonical Wnt factors, such as Wnt5a, which can bind and activate ROR1 on the CLL cell. Wnt5a also can activate NF-κB via ROR1 signaling, which induces production of cytokines, such as IL-6, which can stimulate signal transducer and activator of transcription 3 (STAT3) in the CLL cell or NLCs. NLCs and stromal cells may elaborate Hedgehog (Hh) factors, which interact with surface Pitch 1 (Ptch1) and Ptch2, blocking their capacity to inhibit Smoothened (Smo), allowing for activation of the Hedgehog-signaling pathway in the CLL cell. Cognate interactions between surface cell adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1 or CD106) on the stromal cell, with CLL integrins, such as very late antigen (VLA), enhances CLL cell survival. Self- or environmental antigen (depicted at the bottom of the figure) that is recognized by the sIg expressed by the CLL cell can lead to activation of the BCR signaling pathway. Cognate interactions between the CLL cell and the T cell in the microenvironment also can involve other TNF-family factors, such as CD40 ligand (CD40L or CD154), which enhance expression of chemokines and cytokines, such as IL-4, that further influence the cellular composition of the microenvironment Collectively, the crosstalk between the CLL cell and the accessory cells within this microenvironment provides the perfect niche to foster leukemia-cell proliferation, forming the so-called proliferation centers within lymphoid tissues of patients with CLL. Adapted from Kipps and Choi80 with permission.
The CLL microenvironment. The main types of accessory cells in the CLL microenvironment are depicted surrounding the CLL B cell, which is located at the center.33 CLL cells enter the lymphoid tissue through HEVs (depicted on the right), which also may elaborate chemokines, such as CCL21 and CCL19, which bind CCR7 and can attract CLL cells. HEVs also produce hyaluronic acid (HA), which can activate CD44 on the CLL cell, as it attaches to HEVs via l-selectin (CD62-L) to egress from the blood (CD62-L). In the bottom left corner is a nurse-like cell (NLC), a lymphoma-associated macrophage (LAM) that secretes a number of chemokines, such as CXCL12 or CXCL13, which bind to CXCR4 or CXCR5, respectively, on the CLL cell, attracting it to the microenvironment. The leukemia cell in turn can elaborate chemokines, such as CCL3 and CCL4, which bind to CCR1 and CCR3 (CCR1/3), or CCL17 and CCL22, which bind CCR4, to recruit T cells (depicted in the bottom right) and monocytes, which are the cells that can differentiate into NLCs. Cognate interactions between CD31 and CD38 on NLCs and CLL cells, respectively, may promote CLL-cell survival. The NLC also expresses proteins of the TNF family, such as BAFF (CD257) and a proliferation inducing ligand (APRIL or CD256), which in turn activate TNF-family receptors such as transmembrane activator and calcium-modulator and cyclophilin ligand (CAML) interactor (TACI or CD267), B-cell maturation antigen (BCMA or CD269), or BAFF-receptor (BAFF-R or CD268) on the CLL cell, inducing activation of nuclear factor κ-light chain B (NF-κB). NLCs and stromal cells (depicted on top) also can produce factors that stimulate the CLL cell, such as various Wingless-related integration site (Wnt) factors, with interact with Frizzled (Frz) receptors, or noncanonical Wnt factors, such as Wnt5a, which can bind and activate ROR1 on the CLL cell. Wnt5a also can activate NF-κB via ROR1 signaling, which induces production of cytokines, such as IL-6, which can stimulate signal transducer and activator of transcription 3 (STAT3) in the CLL cell or NLCs. NLCs and stromal cells may elaborate Hedgehog (Hh) factors, which interact with surface Pitch 1 (Ptch1) and Ptch2, blocking their capacity to inhibit Smoothened (Smo), allowing for activation of the Hedgehog-signaling pathway in the CLL cell. Cognate interactions between surface cell adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1 or CD106) on the stromal cell, with CLL integrins, such as very late antigen (VLA), enhances CLL cell survival. Self- or environmental antigen (depicted at the bottom of the figure) that is recognized by the sIg expressed by the CLL cell can lead to activation of the BCR signaling pathway. Cognate interactions between the CLL cell and the T cell in the microenvironment also can involve other TNF-family factors, such as CD40 ligand (CD40L or CD154), which enhance expression of chemokines and cytokines, such as IL-4, that further influence the cellular composition of the microenvironment Collectively, the crosstalk between the CLL cell and the accessory cells within this microenvironment provides the perfect niche to foster leukemia-cell proliferation, forming the so-called proliferation centers within lymphoid tissues of patients with CLL. Adapted from Kipps and Choi80 with permission.
Activation of NF-κB can induce expression of miR-155, which in turn can enhance BCR signaling and its downstream activation by reducing expression of INPP5D, which encodes SHIP1 phosphatase.83 As CLL cells exit to the blood, they are relatively deprived of environmental antigen, chemokine, and growth/survival factors, thereby reducing activation of NF-κB and lowering expression of miR-155.83 This may amplify anergy due to consequent upregulation of SHIP1 phosphatase83,84 and contribute to the variable recovery in sIgM expression and function.16,62,66
Cytokines produced by T cells, such as interlukin-4 (IL-4), upregulate sIgM, thereby potentially enhancing BCR-signals. Elaboration of various WNT proteins by cells in the microenvironment can activate canonical and noncanonical WNT signaling pathways.85,86 Activation of ROR1 by WNT5a may also activate NF-kB to enhance BCR activation and proliferation,87 as well as promote migration in response to chemokines.88 In part for this reason, high expression of ROR1 on CLL cells is associated with accelerated disease progression and shorter overall survival both in U-CLL and M-CLL.89
Activation of NF-κB in CLL cells can produce cytokines, such as IL-6, which in turn may induce activation of STAT3 in both leukemic and local accessory cells. Activated CLL cells can produce large amounts of IL-10,90 a feature associated with regulatory B cells,91 which intriguingly express a surface phenotype very similar to that of CLL,92,93 while potentially sustaining an autocrine prosurvival loop on the CLL cells. This could have relevance for the immune suppression observed in patients with CLL.93
Hedgehog signaling in response to Desert Hedgehog (DHH) present in the microenvironment may provide pro-survival and growth stimulation for at least a subset of patients,94 particularly those with trisomy 12.95,96 Some cases with trisomy 12 and elevated levels of GLI1 and PTCH1 transcripts have autocrine DHH and may be particularly responsive in vitro to SMOOTHENED (SMO) inhibitors, which were less effective when the CLL cells were cultured on DHH-expressing stroma.96 The capacity of stroma to mitigate that activity of SMO inhibitors, and/or the acquisition of somatic mutations encoding proteins in the Hh p-signaling pathway that are downstream of SMO or PTCH1,94 could account for the apparent lack of clinical response of in the small numbers of patients treated with SMO inhibitors.97
Because expression of CXCR4 is downmodulated in response to CXCL12,98 CLL cells just exiting lymphoid tissues are CXCR4dim and have higher expression of activation-induced CD5 (known as CD5bright cells)65 relative to the CLL cells poised to re-enter lymphoid compartments.99 However cells expressing these 2 extremes form a very low percentage of the CLL cells in the blood, with the majority expressing intermediate levels of both.99 Although variable, a higher level expression of chemokine receptors by transiting CLL cells deprived of their microenvironment incentivizes their migration back into the lymphoid tissues. A rather counterintuitive finding is that the small fraction of the recent emigrants from tissue express higher levels of sIgM.100 This suggests that, in contrast to loss by endocytosis, some influences, possibly including locally derived IL-471,101 and/or autocrine IL-6,87 are upregulating sIgM expression in tissue sites.
The role of T cells
The effect of T cells on CLL cells is likely to be critical, since, by analogy with normal B cells, T-cell help is the determinant of cell fate and would allow escape from anergy.102 However T-cell populations in patients are difficult to investigate, mainly because the profile may be perturbed by reactivation of herpes viruses such as Epstein-Barr virus, in the context of immunosuppression.103 The relatively high level of CD8+ T cells is consistent with this, as is the “exhausted” phenotype, which is a feature of chronic antigenic stimulation. Multiple chronic viral infections, including Epstein-Barr virus, lead to downregulation of T-cell function, accompanied by expression of inhibitory receptors such as PD-1.104 Tumor-specific T cells will be hard to find, since CLL driver antigens are likely to be derived from self. In that case, the majority of antigen-specific (cognate) CD4+ T cells will be either deleted or regulated. Normal naive B cells are less dependent on cognate T-cell help, relying instead on help from innate cells, including invariant natural killer T cells, neutrophils, monocyte-derived cells, and/or dendritic cells.102 It is likely that cells of U-CLL retain this capacity and could be more resourceful in capturing available help for survival and proliferation. Normal post-germinal center B cells are more dependent on cognate T-cell help, which will be limited. Death by antigen awaits memory B cells denied T-cell support.20 Cells of M-CLL could reflect this and may rely more heavily on antiapoptotic mechanisms for survival. While this is speculative and based on analogy with normal B and T cells, it could be an important factor in distinguishing tumor behavior in the 2 subsets. All this begs the question of the role of CD40L, known to be a stimulatory factor for CLL cells in vitro but very difficult to detect in proliferation centers. Moreover, CD40L activates both the canonical and noncanonical NF-κB signaling pathways, while the gene expression profile of CLL isolated from lymph nodes indicates primarily activation of the canonical pathway alone,15 suggesting that receptors other than CD40 contribute to cell activation in vivo.82,87
Autonomous signaling
It is well known that Ig-Ig interactions occur in the B-cell membrane. Oligomerization can now be observed at the nanoscale level with the spontaneous formation of nanoclusters of sIgM and sIgD.105 These can form distinct compartments in the cell membrane that incorporate different groups of proteins and lipids. They include proteins such as CD45 and CD22, which can modulate BCR signaling. Only upon exposure to antigen are these nanoclusters reorganized to allow a calibrated level of signaling and activation.106
It has been proposed that an Ig-Ig interaction in the absence of exogenous antigen drives B-cell responses in CLL.17 This is postulated to occur via interaction between amino acids in HCDR3 and an internal epitope of another Ig molecule. The question that arises concerns the function of this interaction and whether it occurs in vivo in the presence of serum Ig. Even so, a model proposing that CLL is stimulated exclusively via Ig-Ig interactions independent of exogenous antigen cannot readily explain the differences in clinical behavior or Ig-signaling noted for U-CLL versus M-C LL or the ability to reverse anergy of CLL cells that are cultured in vitro.
The most detailed documentation of Ig-Ig effects appears to be for 2 exceptional subsets of CLL: (1) IgG-positive cases expressing IGHV4-34, which interact via IGHVCDR3/FR1 with the CH1 of a second Fab,107 and (2) the unusual cases of IGHV3-21 with the associated IGLV3-21*01 light chain, which can acquire a facilitating mutation at the IGLJ-IGLC border.108 Even there, the interactions are very weak, and although one role might be to create a “readiness to signal,” it remains to be seen if those Ig-Ig interactions alone can influence proliferation or survival in vivo.
Concluding remarks
CLL remains a focus for linking biology to therapy, and the modern dual approaches of targeting the BCR and apoptosis are having a dramatic impact on this tumor. Defining the 2 major subsets provides information on prognosis that can be part of clinical assessment, especially as new genetic techniques for assigning patients are becoming available. However, gaps in our knowledge remain, including the nature of driving self-antigens, T-cell involvement, the control of cell migration through tissues, and how inhibitors impinge on the overall behavior of tumor cells in vivo. Development of therapy resistance generally is inevitable with tumors that are not eradicated by treatment. Understanding the molecular and biologic mechanisms responsible for such resistance is required for the development of salvage therapy or combination therapy more likely to eradicate the neoplastic clone. Transformation to a more aggressive disease continues to be a concern, and gene-based probing is revealing significant changes.109 Opportunities to target CLL at all stages as required will emerge as biology continues to reveal pinch points of vulnerability in the tumor cells.
Acknowledgments
This work was supported by Blood Cancer UK/Bloodwise (grant 18009); the National Institutes of Health, National Cancer Institute (R01-CA236361) (T.J.K.); the Keanu Eyles Hematology Fellowship; the Cancer Research UK ECRIN-M3 program (grant C42023/A29370); the CRUK CLL program (C2750/A23669); and CRUK Southampton Centre core funding (C36811/A29101).
Authorship
Contribution: F.K.S., F.F., and T.J.K. wrote the paper, contributed comments, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Freda K. Stevenson. School of Cancer Sciences, University of Southampton, Southampton, SO16 6YD, United Kingdom; e-mail: fs@soton.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal