

Key Points
Gene therapy with AAV8 codon-optimized human FIX Padua complementary DNA induced FIX expression in hemophilia B patients.
Sustained transgene expression, achieved in only 1 participant, was probably hindered by vector-mediated proinflammatory responses.

Abstract
Gene therapy has the potential to maintain therapeutic blood clotting factor IX (FIX) levels in patients with hemophilia B by delivering a functional human F9 gene into liver cells. This phase 1/2, open-label dose-escalation study investigated BAX 335 (AskBio009, AAV8.sc-TTR-FIXR338Lopt), an adeno-associated virus serotype 8 (AAV8)–based FIX Padua gene therapy, in patients with hemophilia B. This report focuses on 12-month interim analyses of safety, pharmacokinetic variables, effects on FIX activity, and immune responses for dosed participants. Eight adult male participants (aged 20-69 years; range FIX activity, 0.5% to 2.0%) received 1 of 3 BAX 335 IV doses: 2.0 × 1011; 1.0 × 1012; or 3.0 × 1012 vector genomes/kg. Three (37.5%) participants had 4 serious adverse events, all considered unrelated to BAX 335. No serious adverse event led to death. No clinical thrombosis, inhibitors, or other FIX Padua–directed immunity was reported. FIX expression was measurable in 7 of 8 participants; peak FIX activity displayed dose dependence (32.0% to 58.5% in cohort 3). One participant achieved sustained therapeutic FIX activity of ∼20%, without bleeding or replacement therapy, for 4 years; in others, FIX activity was not sustained beyond 5 to 11 weeks. In contrast to some previous studies, corticosteroid treatment did not stabilize FIX activity loss. We hypothesize that the loss of transgene expression could have been caused by stimulation of innate immune responses, including CpG oligodeoxynucleotides introduced into the BAX 335 coding sequence by codon optimization. This trial was registered at www.clinicaltrials.gov as #NCT01687608.
Introduction
Hemophilia B is caused by deficient blood coagulation factor IX (FIX) activity, resulting from variants in the F9 gene.1,2 Gene therapy is a potentially curative approach to achieve and maintain therapeutic FIX levels by delivering functioning human F9 genes into hepatocytes using nonpathogenic adeno-associated virus (AAV) vectors.3-8 A limitation of IV administration of recombinant AAV is the barrier posed by preexisting immunity against AAV resulting from natural exposure to wild-type viruses.3,9-11 Transaminitis and loss of FIX expression have been associated with vector dose–dependent activation of circulating cytotoxic T cells putatively recognizing specific epitopes from the AAV capsid. For some patients with asymptomatic increases in alanine aminotransferase (ALT) levels, observed up to 10 weeks post–AAV vector infusion, corticosteroid treatment reportedly stabilizes levels of circulating transgenic FIX activity in blood.5,6,12
Efforts to reduce the immunogenicity observed in the first liver-directed AAV gene therapy trial for hemophilia B3 have focused on maximizing FIX expression while reducing viral vector exposure.6,11,13-15 Consistent with this approach, the AAV serotype 8 (AAV8)–based FIX gene therapy BAX 335 (Baxalta Inc., a Takeda company [Lexington, MA] and Baxalta Innovations GmbH, a Takeda company [Vienna, Austria]) was designed to incorporate a transgene expressing FIX Padua, a hyperactive FIX variant.6,13,16 This gain-of-function variant (leucine substituted for arginine in position 338) exhibits a fivefold to 10-fold increase in FIX-specific activity relative to the wild-type protein.16,17 BAX 335 development included purification steps to reduce the amount of AAV empty capsid to minimize the potential for vector capsid dose–dependent liver inflammation by exposing patients to a lower total capsid dose per FIX genome delivered.13 Encouraging results in preclinical testing led to the further development of BAX 335 as a potential treatment of hemophilia B.
The aims of the current phase 1/2 clinical study were to investigate the safety and pharmacokinetic variables of BAX 335 gene therapy in patients with hemophilia B to assess the impact of treatment on FIX activity and bleeding rates and to monitor immune responses.
Methods
Clinical study design and participants
This first-in-human, phase 1/2, prospective, multicenter, ongoing, open-label study used a single-arm, ascending-dose design to evaluate the safety and pharmacokinetic variables of BAX 335 (AskBio009, AAV8.sc-TTR-FIXR338Lopt) in adults with hemophilia B. Ethical approval was obtained from the institutional review boards of all clinical sites. Written informed consent was provided by all participants. This trial was registered at www.clinicaltrials.gov as #NCT01687608.
The study included male participants (aged 18-75 years) with hemophilia B (FIX ≤2%; FIX <1% for cohort 1 according to the dose-escalation protocol), >150 exposure days to exogenous FIX concentrates, and either ≥3 bleeding episodes per year requiring FIX replacement or regular use of FIX prophylaxis to prevent bleeding. Participants were excluded if they had evidence of liver disease, active hepatitis infection, poorly controlled HIV, or neutralizing antibodies (NAbs) against AAV8 (titer >1:5). Eligible individuals were enrolled and screened from January 2013 to June 2016 (details of eligibility criteria and laboratory screening assessments are given in the supplemental Methods, available on the Blood Web site).
Vector production and dosing
The development and production of BAX 335 (scAAV8.FIXR338L) have been described previously.13,14 The BAX 335 expression cassette consisted of a self-complementary transgene flanked by AAV2-derived inverted terminal repeats, liver-specific transthyretin promoter/enhancers, and the codon-optimized complementary DNA (cDNA) encoding the hyperactive FIX (R338L) Padua variant (Figure 1; supplemental Figure 1). Codon optimization involves the engineering of conservative mutations in a nucleotide sequence of the gene expression cassette (ie, nucleotide changes that code for the synonymous wild-type amino acid sequence) to increase protein expression.18-21

Schematic drawing of the BAX 335 expression cassette. The recombinant vector genome of BAX 335 is designed to express a codon-optimized FIX Padua (R338L) cDNA from the liver-specific murine transthyretin (TTR) promoter/enhancer. The intron fragment from minute virus of mice (MVM) and the bovine growth hormone (BGH) polyadenylation (pA) sequence help improve expression. The expression cassette is flanked by 1 AAV2 wild-type inverted terminal repeat (ITR) sequence (3′ITR) and 1 mutated AAV2 ITR (MUT 5′ITR) to direct preferential replication and packaging of self-complementary (sc) vector DNA. The overall size of the resulting sc genome is 4806 nucleotides (nt). The BAX 335 nucleotide sequence including ITRs is provided in supplemental Figure 1.
Schematic drawing of the BAX 335 expression cassette. The recombinant vector genome of BAX 335 is designed to express a codon-optimized FIX Padua (R338L) cDNA from the liver-specific murine transthyretin (TTR) promoter/enhancer. The intron fragment from minute virus of mice (MVM) and the bovine growth hormone (BGH) polyadenylation (pA) sequence help improve expression. The expression cassette is flanked by 1 AAV2 wild-type inverted terminal repeat (ITR) sequence (3′ITR) and 1 mutated AAV2 ITR (MUT 5′ITR) to direct preferential replication and packaging of self-complementary (sc) vector DNA. The overall size of the resulting sc genome is 4806 nucleotides (nt). The BAX 335 nucleotide sequence including ITRs is provided in supplemental Figure 1.
BAX 335 was manufactured at the Vector Core Facility (University of North Carolina School of Medicine, Chapel Hill, NC) by using a triple plasmid transfection protocol of suspension HEK293 cells grown in the WAVE bioreactor system (GE Healthcare, Piscataway, NJ). The vector titer was determined by using quantitative polymerase chain reaction (PCR) and dot blot assay, as described previously.22,23 The material produced at clinical scale showed a consistently empty capsid content of ∼30%, as determined by transmission electron cryomicroscopy.
Participants were assigned to receive a single IV infusion of BAX 335 in 1 of 3 ascending dose cohorts: (1) 2.0 × 1011 vector genomes (vg)/kg (participants 1 and 2); (2) 1.0 × 1012 vg/kg (participants 3, 4, 5, and 8); and (3) 3.0 × 1012 vg/kg (participants 6 and 7). Participants could receive standard-of-care hemophilia B treatment (including exogenous FIX for on-demand treatment of bleeding episodes and/or prophylaxis) as required.
Study assessments
The primary objective was to evaluate the safety of BAX 335 by assessment of incidence, severity, time of onset, and duration of adverse events (AEs) and analysis of laboratory data (details regarding primary outcome measures are given in the supplemental Methods). Secondary objectives were: (1) to evaluate BAX 335 pharmacokinetic and pharmacodynamic profiles, including the dose required to achieve stable plasma FIX activity 10% to 40% of normal, the need for exogenous FIX replacement, and the duration that BAX 335 genomes remained detectable in bodily fluids (vector shedding in blood, saliva, urine, stool, and semen); and (2) to assess systemic immune responses (humoral and cellular) to the FIX Padua (R338L) transgene product and to AAV8 capsid proteins, at specified time points post–BAX 335 infusion (secondary outcome measures). Binding antibodies against wild-type FIX and FIX Padua were also assayed. Circulating tumor necrosis factor α (and interleukin 6 [IL-6]) levels in serum were measured before and 24 hours after BAX 335 infusion.
FIX activity was measured at the central laboratory by one-stage assay using a BCS XP automated analyzer (Siemens Healthineers, Erlangen, Germany) and ellagic acid as the activator for the activated partial thromboplastin time assay. This assay formed the basis of the Bethesda inhibitor assays used to examine inhibition of clotting in wild-type FIX-containing plasma and in FIX Padua–containing plasma. A transgene product–specific enzyme-linked immunosorbent assay (ELISA) was developed to quantify plasma FIX Padua protein.24 Frequency and severity of bleeding episodes and use of exogenous FIX products during the study were also compared vs data for the 12-month prestudy period.
Statistical analyses
After participants had completed the 12-month study visit, a planned interim analysis was conducted for individual participants, along with summary descriptive statistics for laboratory data and study outcomes. After 12 months, participants entered the long-term follow-up phases of the study.
Poststudy analyses
Root cause analyses, investigating loss of transgene expression, included a mouse model to compare the adaptive immune response elicited by the CpG-rich BAX 335 vector and an otherwise identical CpG-depleted vector. C57BL/6J mice (Charles River Laboratories, Wilmington, MA) were immunized with the 2 vectors at a dose of 4 × 1012 vg/kg. Four weeks after challenge, AAV8 NAb titers were assessed by using a NAb assay adapted to the mouse25 (supplemental Methods).
Whole-exome sequencing and variant analyses were conducted on participant DNA samples to determine potential explanations for variability of FIX Padua expression between participants (supplemental Methods).
Results
Participant demographics and characteristics at screening
Eight eligible male subjects aged 20 to 69 years (mean ± standard deviation 30.5 ± 16.0 years) with hemophilia B were enrolled into 1 of 3 dose cohorts (Table 1). Seven participants were white, and 1 was black/African American; 3 participants had circulating FIX antigen cross-reactive material (CRM) at enrollment. No participant had serologic evidence of active HIV or hepatitis C virus (HCV) (although 2 participants had anti-HCV antibodies, all were negative for HCV RNA according to PCR). All had undetectable levels of AAV8 NAbs (supplemental Table 1). A circulating peripheral blood mononuclear cell (PBMC) response to AAV8 was detected in participant 7 at screening (supplemental Table 2).
Baseline characteristics of participants
| Characteristic | Cohort 1 (2.0 × 1011 vg/kg) | Cohort 2 (1.0 × 1012 vg/kg) | Cohort 3 (3.0 × 1012 vg/kg) | |||||
|---|---|---|---|---|---|---|---|---|
| Participant no. | 1 | 2 | 3 | 4 | 5 | 8 | 6 | 7 |
| Age at informed consent, y | 25 | 29 | 25 | 21 | 24 | 69 | 31 | 20 |
| F9 mutation HGVS* | p.Ser365Argfs*2 | p.Arg-4Gln; p.Arg43Gln | p.Gln121*; p.Gln167* | p.Tyr325*; p.Tyr371* | P.Arg180Trp | p.Ile214HisfsX8 | p.Lys440Glu | p.I316T |
| Baseline FIX activity at screening,† IU/mL | 0.005 | 0.008 | 0.02 | <0.005 | 0.009 | 0.011 | 0.018 | 0.021 |
| Baseline CRM status | Negative | Positive | Negative | Negative | Positive | Negative | Positive | Negative |
| Prior prophylactic FIX replacement therapy (past 12 mo) | No | Yes | Yes | No | No | Yes | Yes | Yes |
| Annualized bleeding rate | 12 | 12 | 7.2 | 12 | 24 | 0 | 3 | 0 |
| Average no. of FIX infusions per year | 24 | 60 | 48 | 12 | 36 | 72 | 48 | 96 |
| HIV status | Negative | Negative | Negative | Negative | Negative | Negative | Negative | Negative |
| HCV antibody status | Negative | Positive | Negative | Negative | Negative | Positive | Negative | Negative |
| Anti-AAV8 NAb titer | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 |
| Anti-AAV2 NAb titer | <1:5 | <1:5 | 1:10 | 1:5 | <1:5 | <1:5 | <1:5 | <1:5 |
| Characteristic | Cohort 1 (2.0 × 1011 vg/kg) | Cohort 2 (1.0 × 1012 vg/kg) | Cohort 3 (3.0 × 1012 vg/kg) | |||||
|---|---|---|---|---|---|---|---|---|
| Participant no. | 1 | 2 | 3 | 4 | 5 | 8 | 6 | 7 |
| Age at informed consent, y | 25 | 29 | 25 | 21 | 24 | 69 | 31 | 20 |
| F9 mutation HGVS* | p.Ser365Argfs*2 | p.Arg-4Gln; p.Arg43Gln | p.Gln121*; p.Gln167* | p.Tyr325*; p.Tyr371* | P.Arg180Trp | p.Ile214HisfsX8 | p.Lys440Glu | p.I316T |
| Baseline FIX activity at screening,† IU/mL | 0.005 | 0.008 | 0.02 | <0.005 | 0.009 | 0.011 | 0.018 | 0.021 |
| Baseline CRM status | Negative | Positive | Negative | Negative | Positive | Negative | Positive | Negative |
| Prior prophylactic FIX replacement therapy (past 12 mo) | No | Yes | Yes | No | No | Yes | Yes | Yes |
| Annualized bleeding rate | 12 | 12 | 7.2 | 12 | 24 | 0 | 3 | 0 |
| Average no. of FIX infusions per year | 24 | 60 | 48 | 12 | 36 | 72 | 48 | 96 |
| HIV status | Negative | Negative | Negative | Negative | Negative | Negative | Negative | Negative |
| HCV antibody status | Negative | Positive | Negative | Negative | Negative | Positive | Negative | Negative |
| Anti-AAV8 NAb titer | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 | <1:5 |
| Anti-AAV2 NAb titer | <1:5 | <1:5 | 1:10 | 1:5 | <1:5 | <1:5 | <1:5 | <1:5 |
According to Human Genome Variation Society nomenclature.
Retesting of FIX activity at screening was performed to ensure appropriate washout from exogenous FIX, specified in the study protocol as at least 5 days after a dose of exogenous short half-life recombinant FIX or 15 days if the participant was using extended half-life FIX.
Safety assessments
BAX 335 infusion
Participants were monitored for hypersensitivity after receiving the first 10% of the BAX 335 infusion. One participant in the lowest dose cohort reported feeling flushed (grade 1 hypersensitivity reaction), considered as a nonserious AE of mild severity, possibly related to BAX 335; the AE resolved within 30 minutes. No participant had his infusion interrupted. An asymptomatic elevation of serum IL-6 levels was reported in 1 participant in the highest dose cohort between 30 minutes and 8 hours postinfusion, which returned to baseline levels 24 hours postinfusion (supplemental Table 3).
Adverse events
Four serious AEs (SAEs) were reported in 3 participants: rhabdomyolysis, bacterial infection of the tonsil, tonsillar hemorrhage, and squamous cell carcinoma of the tonsil. At the 12-month interim analysis, all SAEs except the tonsillar carcinoma were resolved (Table 2). BAX 335–specific quantitative PCR and integration site analyses by nonrestrictive linear amplification-mediated PCR (supplemental Methods) showed no evidence of AAV vector genomes in and/or integration into tonsillar tumor tissue. All SAEs were therefore considered unrelated to BAX 335. The single SAE ongoing at the time of the interim analysis (tonsillar carcinoma) has since been resolved. Ten AEs in 4 participants were considered possibly related to the study treatment (cohort 1, seven AEs in 2 participants; cohort 2, two AEs in 1 participant; and cohort 3, one AE in 1 participant); 7 AEs were mild in severity and 3 were moderate. In cohort 1, there were 5 mild AEs (fatigue, feeling flushed, headache, influenza-like symptoms, and ankle swelling), and 2 were moderate in severity (elevated liver enzyme levels and fatigue); in cohort 2, there was 1 mild AE (high blood pressure) and 1 moderate AE (abscess); and in cohort 3, there was 1 mild AE (elevated liver enzyme levels). At the 12-month interim analysis, 7 of these 10 AEs were resolved, and 3 AEs in 1 participant (cohort 1) were ongoing (ankle swelling, fatigue, and elevated liver enzymes). One of these 3 AEs ongoing at the time of the interim analysis (elevated liver enzyme levels) has since been resolved.
AEs
| Variable | Cohort 1 (2.0 × 1011 vg/kg) | Cohort 2 (1.0 × 1012 vg/kg) | Cohort 3 (3.0 × 1012 vg/kg) | |||||
|---|---|---|---|---|---|---|---|---|
| Participant no. | 1 | 2 | 3 | 4 | 5 | 8 | 6 | 7 |
| AE possibly related to BAX 335, n | 1 | 6 | 0 | 0 | 0 | 2 | 1 | 0 |
| SAE unrelated* to BAX 335, n | 1 | NA | 0 | 2 | 0 | 1 | 0 | 0 |
| AEs possibly related to BAX 335, description (severity) | Tiredness (mild) |
| NA | NA | NA |
| Elevated liver enzyme levels (mild) | NA |
| SAEs unrelated* to BAX 335, description (severity) | Exercise-induced rhabdomyolysis (severe) | NA | NA | Bacterial infection of right tonsil (moderate)Tonsillar hemorrhage (moderate) | NA | Tonsillar carcinoma (severe) | NA | NA |
| Variable | Cohort 1 (2.0 × 1011 vg/kg) | Cohort 2 (1.0 × 1012 vg/kg) | Cohort 3 (3.0 × 1012 vg/kg) | |||||
|---|---|---|---|---|---|---|---|---|
| Participant no. | 1 | 2 | 3 | 4 | 5 | 8 | 6 | 7 |
| AE possibly related to BAX 335, n | 1 | 6 | 0 | 0 | 0 | 2 | 1 | 0 |
| SAE unrelated* to BAX 335, n | 1 | NA | 0 | 2 | 0 | 1 | 0 | 0 |
| AEs possibly related to BAX 335, description (severity) | Tiredness (mild) |
| NA | NA | NA |
| Elevated liver enzyme levels (mild) | NA |
| SAEs unrelated* to BAX 335, description (severity) | Exercise-induced rhabdomyolysis (severe) | NA | NA | Bacterial infection of right tonsil (moderate)Tonsillar hemorrhage (moderate) | NA | Tonsillar carcinoma (severe) | NA | NA |
NA, not applicable.
No SAEs were considered related to BAX 335 treatment.
Vector shedding
Vector genomes were measured in urine, semen, saliva, stool, and whole blood, with peak concentrations and duration of shedding broadly displaying dose dependence across the 3 cohorts (supplemental Table 4). Vector shedding in saliva, urine, semen, or stool was not observed after week 5 in any participant. Vector signal from whole blood was lost in cohorts 1 and 2, remaining above the detection limit at the 12-month visit in the highest dose cohort.
FIX expression
With the exception of the first participant in cohort 1, all participants exhibited vector-derived FIX expression (Figures 2 and 3; supplemental Figure 2) with no evidence of thrombogenicity or cellular and humoral immune responses to FIX Padua. BAX 335–derived FIX expression was dose dependent (Figure 4) and occurred as early as 1 to 2 weeks after BAX 335 infusion in the higher dose cohorts (Figures 2 and 3; supplemental Figure 2). Mean (range) peak FIX activity (for the first 6 months after BAX 335 treatment, excluding values within 5 days of an exogenous FIX infusion) was 2.8% (2.7% to 2.8%) in cohort 1, 12.8% (3.0% to 26.2%) in cohort 2, and 45.3% (32.0% to 58.5%) in cohort 3. At the time of peak expression, circulating coagulation activity derived from gain-of-function FIX Padua (rather than wild-type FIX) was indicated by the FIX-specific activity in participants who were FIX CRM–negative at baseline. Using a FIX Padua–specific ELISA24 corroborated transgene-specific expression for CRM-positive and CRM-negative participants, as the FIX Padua antigen levels correlated with the measured FIX activities but not with the FIX antigen levels measured by using a standard FIX ELISA assay.24
![FIX activity, FIX Padua levels, liver enzymes, and PBMC-mediated immune responses to AAV8 post–BAX 335 infusion for participants in dose cohort 2. FIX activity and FIX Padua activity levels post–BAX 335 infusion by participant in relation to bleeding episodes, administration of FIX replacement therapy, and prednisone dosing (participant 8) as well as liver enzyme markers of hepatotoxicity (ALT and aspartate aminotransferase [AST]) are shown. Study week represents nominal collection time (mean values used if ≥1 result in 1 week). The number of exogenous FIX infusions or bleeding episodes within 1 week is indicated. The lower limit of detection (LOD) for FIX Padua–specific activity (<0.020 U) is shown by the gray dashed line. Participant 8 received prophylactic treatment with prednisone. The duration of prednisone treatment is shown in the green bar chart, with the slope indicating dose tapering. The results from IFN-γ ELISpot assays are shown in the lower graph panel for each participant; the reaction of the participant’s PBMCs to AAV8 capsid peptides in 3 separate pools are plotted as the number of spots per million PBMCs. Values were considered positive if they were above the media control (background) by a factor of 2 and were >60 spots per million PBMCs (black dashed line). Maximum results across all pools were presented for each time point.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/6/10.1182_blood.2019004625/3/m_bloodbld2019004625f2.png?Expires=1767701145&Signature=g9KZxDU2-91MzWi58s6w5WfGL3Ppoo0Rty488SPG1SnJJ-WWiDReklastZboWRVRby-hmvoV3C4wMxkeYIWQWD30ZMbcbOILagcFBbR37oJZxnLz5ntllxlUyYpb~W43AOduUsGY-9GbfMD~db7pMwLFJM3RU1GQPU-NfgsUnJV7434FA9-ckT2FiwcYCOPHFgD01-GOaceQGXHYAPLh4Usr49J~yznt458GLToFZYIdYwIfovjbIqC102RJdtsUMlnri~aYXvoiam6I6-X9bpVvCSL~R3cor7vJxXZQTALPwZcHeCGExRHJmkvRqGeC9GuXcKM7NgQxoPAv0Uq-Jw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FIX activity, FIX Padua levels, liver enzymes, and PBMC-mediated immune responses to AAV8 post–BAX 335 infusion for participants in dose cohort 2. FIX activity and FIX Padua activity levels post–BAX 335 infusion by participant in relation to bleeding episodes, administration of FIX replacement therapy, and prednisone dosing (participant 8) as well as liver enzyme markers of hepatotoxicity (ALT and aspartate aminotransferase [AST]) are shown. Study week represents nominal collection time (mean values used if ≥1 result in 1 week). The number of exogenous FIX infusions or bleeding episodes within 1 week is indicated. The lower limit of detection (LOD) for FIX Padua–specific activity (<0.020 U) is shown by the gray dashed line. Participant 8 received prophylactic treatment with prednisone. The duration of prednisone treatment is shown in the green bar chart, with the slope indicating dose tapering. The results from IFN-γ ELISpot assays are shown in the lower graph panel for each participant; the reaction of the participant’s PBMCs to AAV8 capsid peptides in 3 separate pools are plotted as the number of spots per million PBMCs. Values were considered positive if they were above the media control (background) by a factor of 2 and were >60 spots per million PBMCs (black dashed line). Maximum results across all pools were presented for each time point.
FIX activity, FIX Padua levels, liver enzymes, and PBMC-mediated immune responses to AAV8 post–BAX 335 infusion for participants in dose cohort 2. FIX activity and FIX Padua activity levels post–BAX 335 infusion by participant in relation to bleeding episodes, administration of FIX replacement therapy, and prednisone dosing (participant 8) as well as liver enzyme markers of hepatotoxicity (ALT and aspartate aminotransferase [AST]) are shown. Study week represents nominal collection time (mean values used if ≥1 result in 1 week). The number of exogenous FIX infusions or bleeding episodes within 1 week is indicated. The lower limit of detection (LOD) for FIX Padua–specific activity (<0.020 U) is shown by the gray dashed line. Participant 8 received prophylactic treatment with prednisone. The duration of prednisone treatment is shown in the green bar chart, with the slope indicating dose tapering. The results from IFN-γ ELISpot assays are shown in the lower graph panel for each participant; the reaction of the participant’s PBMCs to AAV8 capsid peptides in 3 separate pools are plotted as the number of spots per million PBMCs. Values were considered positive if they were above the media control (background) by a factor of 2 and were >60 spots per million PBMCs (black dashed line). Maximum results across all pools were presented for each time point.
![FIX activity, FIX Padua levels, liver enzyme levels, and PBMC-mediated immune responses to AAV8 post–BAX 335 infusion for participants in dose cohort 3. FIX and FIX Padua activity post–BAX 335 infusion by participant in relation to bleeding episodes, administration of FIX replacement therapy, and prednisone dosing (participants 6 and 7) as well as liver enzyme markers of hepatotoxicity (ALT and aspartate aminotransferase [AST]) are shown. Study week represents nominal collection time (mean values used if ≥1 result in 1 week). The number of exogenous FIX infusions or bleeding episodes within 1 week is indicated. Participants 6 and 7 received prednisone as a rescue treatment in response to a sudden loss of FIX expression. The duration of prednisone treatment is shown in the green bar charts, with the slope indicating dose tapering. FIX Padua–specific activity values were above the lower limit of detection (LOD) (<0.020 U), which is shown by the gray dashed line. The results from IFN-γ ELISpot assays are shown in the lower graph panel for each participant; the reaction of the participant’s PBMCs to AAV8 capsid peptides in 3 separate pools are plotted as the number of spots per million PBMCs. Values were considered positive if they were above the media control (background) by a factor of 2 and were >60 spots per million PBMCs (black dashed line). Maximum results across all pools were presented for each time point.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/6/10.1182_blood.2019004625/3/m_bloodbld2019004625f3.png?Expires=1767701145&Signature=bCpVkXDFhDzpmqpgeEIC8KZErSapYhBq5a1z0nRE4RvJyFpvt9LacWSkE2L6vL9he6Vea4GYiNMY85mUpC~KntC-xQrwxz6c~Y4LGyE3t20di4Un1CF2ufj50luXdzp0Hr9IdSp~eJDQqzP4LKo1cafGG4sqRTjguBqRwuAHZ5Zh79tRKA61nDGtDZI7QdYMggD0u2-WeGXKBDesB-GYlyo8m7ulD83aln7gWjp7vETY-emybDIJ87vxFIk3UvStoYuHTknyfkw-YxMuIKW67~QEQpisYOY80XP3wmsVNv85kYBIeUHzkzTrN0b7aRW7lHiISjV6mky603YawcbvkQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FIX activity, FIX Padua levels, liver enzyme levels, and PBMC-mediated immune responses to AAV8 post–BAX 335 infusion for participants in dose cohort 3. FIX and FIX Padua activity post–BAX 335 infusion by participant in relation to bleeding episodes, administration of FIX replacement therapy, and prednisone dosing (participants 6 and 7) as well as liver enzyme markers of hepatotoxicity (ALT and aspartate aminotransferase [AST]) are shown. Study week represents nominal collection time (mean values used if ≥1 result in 1 week). The number of exogenous FIX infusions or bleeding episodes within 1 week is indicated. Participants 6 and 7 received prednisone as a rescue treatment in response to a sudden loss of FIX expression. The duration of prednisone treatment is shown in the green bar charts, with the slope indicating dose tapering. FIX Padua–specific activity values were above the lower limit of detection (LOD) (<0.020 U), which is shown by the gray dashed line. The results from IFN-γ ELISpot assays are shown in the lower graph panel for each participant; the reaction of the participant’s PBMCs to AAV8 capsid peptides in 3 separate pools are plotted as the number of spots per million PBMCs. Values were considered positive if they were above the media control (background) by a factor of 2 and were >60 spots per million PBMCs (black dashed line). Maximum results across all pools were presented for each time point.
FIX activity, FIX Padua levels, liver enzyme levels, and PBMC-mediated immune responses to AAV8 post–BAX 335 infusion for participants in dose cohort 3. FIX and FIX Padua activity post–BAX 335 infusion by participant in relation to bleeding episodes, administration of FIX replacement therapy, and prednisone dosing (participants 6 and 7) as well as liver enzyme markers of hepatotoxicity (ALT and aspartate aminotransferase [AST]) are shown. Study week represents nominal collection time (mean values used if ≥1 result in 1 week). The number of exogenous FIX infusions or bleeding episodes within 1 week is indicated. Participants 6 and 7 received prednisone as a rescue treatment in response to a sudden loss of FIX expression. The duration of prednisone treatment is shown in the green bar charts, with the slope indicating dose tapering. FIX Padua–specific activity values were above the lower limit of detection (LOD) (<0.020 U), which is shown by the gray dashed line. The results from IFN-γ ELISpot assays are shown in the lower graph panel for each participant; the reaction of the participant’s PBMCs to AAV8 capsid peptides in 3 separate pools are plotted as the number of spots per million PBMCs. Values were considered positive if they were above the media control (background) by a factor of 2 and were >60 spots per million PBMCs (black dashed line). Maximum results across all pools were presented for each time point.
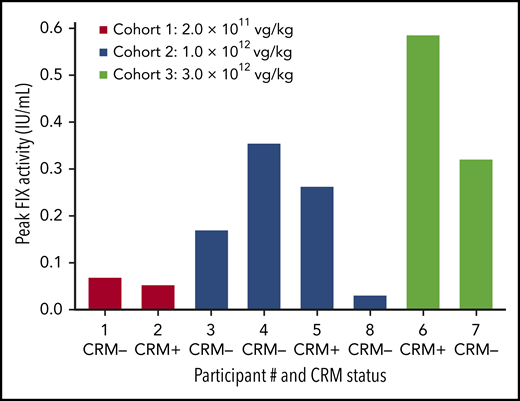
Peak FIX activity by participant, baseline CRM status, and dose cohort. FIX activity was measured in each participant during the 6 months after BAX 335 infusion. Participants, testing either positive or negative for circulating FIX antigen CRM at baseline, received a single infusion of BAX 335 at 1 of 3 doses. FIX measurements were determined by one-stage clotting assay (central laboratory), including measurements within 5 days of an exogenous FIX infusion and measurements from unscheduled study visits.
Peak FIX activity by participant, baseline CRM status, and dose cohort. FIX activity was measured in each participant during the 6 months after BAX 335 infusion. Participants, testing either positive or negative for circulating FIX antigen CRM at baseline, received a single infusion of BAX 335 at 1 of 3 doses. FIX measurements were determined by one-stage clotting assay (central laboratory), including measurements within 5 days of an exogenous FIX infusion and measurements from unscheduled study visits.
In cohort 1, participant 2 achieved ∼4% FIX activity at week 2 (supplemental Figure 2); peak FIX activity was 2.7% when excluding FIX activity data within 5 days of exogenous FIX infusion. All 4 participants in cohort 2 experienced significant levels of FIX activity; however, there was considerable inter-participant variability (Figure 2). Participant 3 exhibited FIX activity with a peak of 16.9% at week 11 (14.6% excluding data within 5 days of exogenous FIX infusion). This peak was followed by an acute loss of FIX expression at week 12, and the patient resumed prophylactic FIX infusions. Participant 4 had an initial peak FIX activity of 7.3% (excluding data within 5 days of exogenous FIX infusion), which subsequently fell between weeks 6 and 7; he also resumed replacement FIX therapy for bleed treatment. Participant 8 received prophylactic corticosteroid treatment; FIX activity of 1% to 3% was attained for the first 6 months (peak 3.0% excluding data within 5 days of exogenous FIX infusion). Participant 5 achieved FIX activity of ∼20% at week 7 and was the only participant with sustained activity (achieved in the absence of bleeding or FIX protein infusions) from week 2 to 52 postinfusion; FIX activity was sustained during 4 years’ follow-up. Although participants 3 and 4 lost >80% of peak expression acutely (over ∼2 weeks), neither exhibited increased liver transaminase levels, vector- or transgene-specific T-cell responses, development of inhibitors or nonneutralizing antibodies to FIX or to FIX Padua, or other symptoms or laboratory perturbations coincident with this loss.
In the highest dose cohort, participant 6 achieved exogenous FIX-corrected peak FIX activity of 58.5% by week 6, when an increase in AAV capsid–directed T-cell activation was observed. A steep fall in FIX activity occurred by week 7, accompanied by an increase in liver transaminase levels and further T-cell activation. Per-protocol treatment with prednisone was initiated within 72 hours, resulting in rapid normalization of liver enzyme levels and interferon γ (IFN-γ) ELISpot results. FIX activity did not stabilize, and the participant resumed prophylactic FIX infusions. Participant 7 exceeded 30% FIX activity by week 5 (exogenous FIX-corrected peak FIX activity of 32.0%). He was started on prednisone after experiencing a slight elevation in liver enzyme levels together with a sudden loss of FIX activity at week 6; however, FIX activity continued to decline (Figure 3).
AAV8-specific T-cell responses against the vector
It is hypothesized that CD8 cytotoxic T cells could kill transduced hepatocytes and might be responsible for the decline in transgene expression.3,26 However, only 2 participants, both in the highest dose cohort, had strong IFN-γ ELISpot signals for circulating AAV8 capsid–reactive T cells; ALT/aspartate aminotransferase levels were elevated in these patients (Figure 3). Although corticosteroid therapy was associated with immediate normalization of the IFN-γ ELISpot in participant 6, this signal remained elevated for weeks after the initiation of prednisone in participant 7.
Systemic corticosteroid administration initiated in response to ALT elevations in participants 6 and 7, and as prophylaxis in participant 8, did not stabilize FIX activity levels in these participants (Figure 3).
Poststudy analysis: investigation into the acute loss of FIX Padua expression
Analysis of cytokines and liver parameters did not provide any insight into why only participant 5 exhibited stable FIX Padua expression for 4 years.
Recent studies in mouse models report that vectorized expression cassettes harboring CpG motifs may activate the innate immune system via Toll-like receptor 9 (TLR9), with a potentially negative impact on transgene expression via supporting cell–mediated and/or antibody-mediated immunity toward AAV vector–associated antigens.27,28 Before codon optimization, native FIX and FIX Padua cDNA sequences each contained 19 CpG motifs. The original GeneArt codon optimization algorithm used to generate the BAX 335 FIX Padua cDNA addressed the understanding that gene sequences with greater GC content lead to higher messenger RNA levels and greater protein translation vs those with greater AT content.19,29 As reported by Wu et al,14 FIX codon optimization incorporated in BAX 335 led to increased FIX expression in mouse liver vs wild-type FIX cDNA. Nevertheless, the optimization algorithm resulted in a FIX Padua transgene in BAX 335 containing elevated CpG density (99 CpG motifs) and CpG clusters (supplemental Figure 1). In an animal model in which vector-dependent activation of the innate immune system was analyzed indirectly by measuring the titers of NAbs against AAV8, mice were challenged with BAX 335 or a BAX 335–like construct bearing a FIX Padua transgene that was codon-optimized to deplete CpG motifs. NAb titers against AAV8 were increased in mice treated with BAX 335 (Figure 5).

Anti-AAV8 NAb responses in mice treated with BAX 335 vs CpG-depleted vector constructs. Anti-AAV8 NAb responses, as a surrogate marker for immune activation via TLR9, indicate lower immunogenicity for CpG-depleted vectors. C57BL/6J mice (8-10 weeks old, n = 6-8 per group) were treated IV with identical doses (4 × 1012 vg/kg) of either one of two AAV8 vectors carrying an FIX Padua coding sequence with different numbers of CpG oligodeoxynucleotides (ODNs): BAX 335 (99 CpG ODNs); CpG-depleted candidate (0 CpG ODNs). Blood was collected 4 weeks later, and the magnitude of the resulting titer of anti-AAV8 NAbs was assayed as a marker of adaptive immune responses to CpG ODNs.
Anti-AAV8 NAb responses in mice treated with BAX 335 vs CpG-depleted vector constructs. Anti-AAV8 NAb responses, as a surrogate marker for immune activation via TLR9, indicate lower immunogenicity for CpG-depleted vectors. C57BL/6J mice (8-10 weeks old, n = 6-8 per group) were treated IV with identical doses (4 × 1012 vg/kg) of either one of two AAV8 vectors carrying an FIX Padua coding sequence with different numbers of CpG oligodeoxynucleotides (ODNs): BAX 335 (99 CpG ODNs); CpG-depleted candidate (0 CpG ODNs). Blood was collected 4 weeks later, and the magnitude of the resulting titer of anti-AAV8 NAbs was assayed as a marker of adaptive immune responses to CpG ODNs.
We further hypothesized that a genetic variation specific to the individual could have contributed to the failure of participant 5 to mount an anti-AAV8 immune response, facilitating the long-term transgene expression observed in this participant. We therefore investigated genetic variations in all participants by whole-exome sequencing. There were no homozygous gene variants unique to the genome of participant 5. Unique heterozygous and compound heterozygous variants were found. The most indicative variant was a heterozygous missense variant c.344A>C, p.A115E in the IL6R gene encoding the receptor for IL-6, causing an alanine-for-glutamate substitution within the IL-6–binding domain predicted to adversely affect receptor function (Combined Annotation Dependent Depletion score of 26.1).30 Furthermore, haploinsufficiency of IL-6R in humans and mice has been documented.31-33
Discussion
AAV8-based FIX Padua BAX 335 gene therapy was well tolerated in all 8 adult participants of this phase 1/2 study. There were no notable infusion-related or subsequent safety abnormalities, no thrombosis, and no evidence of inhibitors or other FIX Padua–directed immunity during the 1-year study or subsequent follow-up. Treatment with BAX 335 resulted in gene transfer of the codon-optimized FIX Padua transgene in liver cells and expression of hemostatically functional FIX in 7 of 8 participants. BAX 335 administration was associated with dose-dependent increases in peak FIX activity unequivocally caused by expression of the transgene product (Figure 2). Functional FIX Padua expression resulted in short-term reductions in factor consumption (B.E., P.E.M., unpublished data, 26 April 2017), consistent with a study of patients with hemophilia B receiving a different FIX Padua vector.6 Peak FIX activity levels were not sustained in 6 of 7 participants with measurable FIX expression. In the 1 participant who achieved long-term FIX expression, a therapeutic FIX activity of ∼20% has been observed for 4 years without bleeding or use of FIX replacement therapy.
Four SAEs reported in 3 participants, including tonsillar carcinoma, were considered unrelated to BAX 335 treatment. BAX 335–specific quantitative PCR and integration site analyses on tonsillar tumor biopsy tissue found no evidence of AAV vector genomes in and/or integration into tonsillar tumor tissue. In support of this finding, genome sequences putatively associated with hepatocellular carcinoma risk in a previous nonclinical study of wild-type AAV integration were examined, and they exhibited no integrated vector sequences or other disruption.34,35
Acute loss of transgene expression within the first few months after infusion, often accompanied by elevation of liver enzyme levels (exemplified by participant 6), has also been reported in other AAV vector hemophilia gene therapy trials.3,5-7,12,15,36 This clinically asymptomatic hepatotoxicity, coincident with acute loss in transgene expression, has been attributed to a vector dose–dependent capsid-directed cellular immune response.3,26 The strongest IFN-γ ELISpot and transaminase signals were measured in the highest BAX 335 dose cohort and support the relationship to vector dose evident from previous trials.15
In contrast to some other AAV FIX gene therapy studies, in which capsid-directed adaptive immune responses limiting FIX expression could be stabilized by immunosuppression,5,6,12 FIX activity could not be stabilized by corticosteroid rescue or prophylaxis in the current study. An AAV8 FIX gene therapy study at the Children’s Hospital of Philadelphia (#NCT01620801) similarly reported 2 of 3 patients with vector capsid immune responses that could not be stabilized by immunosuppression.37 Thus, a clear association between corticosteroid immunosuppression and the ability to normalize ALT levels and stabilize factor expression has not been shown in all patients receiving AAV gene therapy for hemophilia; the basis of this variability has yet to be elucidated.
The root cause for the loss of expression in all but one participant is unknown. We cannot exclude manufacturing-related causes such as vector purity or the full:empty capsid particle ratio of the product. In this study, the triple transfection protocol for AAV production developed by Grieger et al23 was used. This protocol has now been used for numerous clinical studies (eg, Canavan disease, giant axonal neuropathy, Duchenne muscular dystrophy), including generating phase 3 Duchenne muscular dystrophy material without similar incident, strongly suggesting clinical observations from this study may derive from elsewhere. However, sequence-related outcomes affecting either DNA vector genome structure or function are more likely. In this case, codon optimization of the FIX Padua transgene accidentally introduced into the BAX 335 vector genome clusters of CpG motifs, which are pathogen-associated molecular patterns known to stimulate innate immune responses through TLR9.38,39 We therefore reasoned that CpG enrichment could serve as a plausible explanation. Exploring this hypothesis further, emulating these responses nonclinically is difficult because animal models of liver-directed AAV gene therapy40-43 do not predict or reproduce the cell-mediated immune response directed against AAV and the loss of transduced hepatocytes reported clinically.4,13,44 Thus, the validity of these animal models for assessing the comparative immunogenicity of similar FIX-expressing vectors is unclear. Nevertheless, innate immune responses against recombinant AAV may potentially augment the activation of adaptive T-cell and B-cell immune responses.37,39,45 To test CpG-mediated pathogen-associated molecular pattern stimulation of innate immune responses, we compared the strength of the humoral adaptive immune response elicited by BAX 335 and an otherwise comparable but CpG-reduced vector as a marker of innate immune activation. Higher AAV8 NAb titers developed in response to BAX 335, suggesting that the increased number of CpG motifs in the clinical vector may have contributed to the irreversible loss of FIX expression. Although the data from this study and animal studies support a strong correlation between CpG motifs and clinical outcome, recent animal studies have unraveled additional innate immune interactions between AAV and host that may be at play and cannot be ignored.46
Loss of FIX expression in participants 6 and 7, receiving the highest BAX 335 dose, was concurrent with an increase in liver enzyme levels and PBMCs reactive to AAV8 (measured by IFN-γ ELISpot). However, this was not the case in the lower dose cohort for participants 3 and 4. Relatively weak immune responses to the vector localized in the liver might not always be detectable in assays of peripheral blood.
Transient elevation of IL-6 as an indicator of innate immune system activation has been described in animal studies mapping TLR9-dependent activation of NF-κB and transient proinflammatory cytokine induction after liver-directed infusion of AAV vector47 and in AAV-exposed PBMCs.48 Transient induction of the proinflammatory cytokine IL-6 after infusion of the highest BAX 335 dose in participant 6 (supplemental Table 3) may be further evidence of an innate immune response triggered by recombinant AAV in humans. The heterozygous genetic variant in the IL6R gene of participant 5, identified by sequence analysis, has a Combined Annotation Dependent Depletion score30 indicating a high probability of deleteriousness to the function of the IL6R gene product. This potential IL-6 receptor haploinsufficiency is suggestive of participant 5 being less susceptible to innate immune signaling driven by IL-6–mediated inflammatory stress caused by an anti-AAV immune response. Although the p.A115E variant identified in participant 5 has not been studied, there is precedent for a nonsynonymous IL6R single-nucleotide polymorphism to be associated with reduced functionality and impaired IL-6 responsiveness.31 This supports the notion that although participant 5 might not display the clinical phenotype of hypofunctional immunity under homeostatic conditions, when presented with a strong immune adjuvant, the immune response might be incomplete.
Previous clinical trial experience has implicated adaptive immune responses directed at the capsid as the main factor limiting sustained expression from AAV vectors. Here, we suggest that innate immune stimulation, potentially driven by CpG enrichment (introduced during codon optimization of the gene expression cassette) could have contributed to the loss of AAV vector–mediated gene expression in a human clinical trial. Although future research will need to substantiate this hypothesis, avoiding potential immune stimuli within the nucleotide sequence is increasingly recognized as an important general consideration for the design of DNA gene therapy vectors and has led to the development of CpG-reduced vectors for gene therapy. Implications for future clinical studies of any systemically delivered AAV vectors include increased vigilance for immunostimulatory components introduced during vector production,49 vector codon optimization and design, and early control of innate immunity to reduce vector immunogenicity and improve AAV transduction efficiency. Success in controlling innate immunity may also be influenced by improved understanding of the impact of interindividual variations underlying immune responses, including in genes that are yet to be identified.
The data sets, including the redacted study protocol, redacted statistical analysis plan, and individual participants’ data supporting the results reported in this article, will be made available within 3 months from initial request (www.takeda.com/what-we-do/research-and-development/takeda-clinical-trial-transparency/) to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the BAX 335 study participants and their families, as well as the study investigators and study contributors, including the following study investigators who contributed with screening, enrollment, and/or long-term follow-up of study participants: Jonathan Ducore, Shawn Jobe, Doris Quon, Michael Recht, and Mark Reding. They also thank Jade J. Samulski for support of early preclinical and clinical program development (Asklepios BioPharmaceutical), and Alfred Weber (FIX Padua ELISA, Baxalta Innovations GmbH, a Takeda company), Marco Frentsch (murine immunogenicity studies, BIH Center for Regenerative Therapies), Arthur R. Thompson (Data and Safety Monitoring Board chair), and Edward Gomperts (clinical trial medical monitor).
This research was funded by Baxalta US Inc., a member of the Takeda group of companies, and Baxalta Innovations GmbH, a member of the Takeda group of companies. The project described was supported in part by grant RC3HL103396 from the National Institutes of Health, National Heart, Lung, and Blood Institute to S.W.J.M. (co–Principal Investigator) and P.E.M. (co–Principal Investigator). Medical writing support for the manuscript was provided by Isobel Lever, employee of Excel Medical Affairs, and was funded by Baxalta US Inc., a member of the Takeda group of companies.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Authorship
Contribution: P.E.M. designed and executed the study, contributed to data analysis and interpretation, wrote the first draft of the manuscript, and significantly contributed to the writing and editing of subsequent drafts; S.W.J.M. contributed to the design of the study and data interpretation, and edited the manuscript; R.J.S. contributed to design of the original BAX 335 vector, vector production and testing in the first 3 patients, data analysis and interpretation, and critical revision of the final manuscript; B.M.R. contributed to the design of immunological analysis, data collection and interpretation, and writing and editing of the manuscript; H.R. contributed to data collection, data analysis and interpretation, and significantly contributed to writing and editing of the manuscript; M.d.l.R. designed experiments to assess the impact of CpGs and contributed to data analysis and interpretation, and writing and editing of the manuscript; J.C.C., B.E., I.B., and F.S. contributed to data collection, data analysis and interpretation, and writing and editing of the manuscript; and B.A.K., C.E.W., M.A.E., N.C.J., G.Y., and A.v.D. contributed to data collection and interpretation, and edited the manuscript.
Conflict-of-interest disclosure: B.A.K. has received research funding from Bioverativ, Pfizer, Sangamo, Shire (a Takeda company), and Spark Therapeutics; and has received payment for consulting from BioMarin, Pfizer, Sanofi, and Spark Therapeutics. M.A.E. has received honoraria and consulting fees from Baxalta, a member of the Takeda group of companies, Bayer, CSL Behring, Genentech/Roche, Kedrion, Novo Nordisk, Pfizer, and Sanofi. G.Y. has received honoraria and consulting fees from Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Pfizer, Shire, a member of the Takeda group of companies, Spark, and UniQure. A.V.D. has received honoraria and consulting fees from Baxalta, Bayer, BioMarin, Bioverativ/Sanofi, CSL Behring, Hema Biologics, Novo Nordisk, Pfizer, Shire, a member of the Takeda group of companies, and uniQure; and is cofounder and member of the board of directors of Hematherix LLC., a biotech company that is developing superFV, a therapy for bleeding complications. S.W.J.M. was an employee of Asklepios and a co-owner of Chatham at the time of the study and may receive remuneration from future development of this program. R.J.S. is the founder and a shareholder at Asklepios BioPharmaceutical and Bamboo Therapeutics, Inc.; holds patents that have been licensed by the University of North Carolina to Asklepios BioPharmaceutical, for which he receives royalties; and has consulted for Baxter Healthcare and has received payment for speaking. I.B. is an employee of Baxalta Innovations GmbH, a member of the Takeda group of companies. M.d.l.R. was an employee of Baxalta Innovations GmbH, a member of the Takeda group of companies, at the time of the study. B.M.R. was an employee of Baxalta Innovations GmbH, a member of the Takeda group of companies, at the time of the study and is a Takeda stock owner. H.R. was an employee of Baxalta Innovations GmbH, a member of the Takeda group of companies, at the time of the study and is a Takeda stock owner. F.S. was an employee of Baxalta Innovations GmbH, a member of the Takeda group of companies, at the time of the study and is a Takeda stock owner. J.C.C. is an employee of Baxalta US Inc., a member of the Takeda group of companies, and is a Takeda stock owner. B.E. is an employee of Baxalta US Inc., a member of the Takeda group of companies. P.E.M. was an employee of the University of North Carolina and was subsequently an employee of Baxalta US Inc., a member of the Takeda group of companies, at the time of the study (with no conflicts to disclose related to this analysis); he holds patents that have been licensed to Asklepios BioPharmaceutical, for which he receives royalties; and he has received research support through the University of North Carolina from Asklepios BioPharmaceutical, Baxter Healthcare, and Novo Nordisk. The remaining authors declare no competing financial interests.
The current affiliation for M.d.l.R. is Sangamo Therapeutics, Brisbane, CA.
The current affiliation for B.M.R. is IMC University of Applied Sciences Krems, Vienna, Austria.
The current affiliation for H.R. is Evotec GT GmbH, Orth an der Donau, Austria.
The current affiliation for F.S. is Evotec GT GmbH, Orth an der Donau, Austria.
The current affiliation for P.E.M. is Spark Therapeutics, Inc. Chapel Hill, NC.
Correspondence: Bruce Ewenstein, Baxalta US Inc., a member of the Takeda group of companies, 650 E Kendall St, Cambridge, MA 02142; e-mail: bruce.ewenstein@takeda.com.
