

Key Points
aHUS-associated FHR-1 mutants are pathogenic because they acquire capacity to bind sialic acids, which allows C3b-binding competition with FH.
The mechanism by which surface-bound FHR-1 promotes complement activation is the binding and attraction of native C3 to the cell surface.

Abstract
Factor H (FH)–related proteins are a group of partly characterized complement proteins thought to promote complement activation by competing with FH in binding to surface-bound C3b. Among them, FH-related protein 1 (FHR-1) is remarkable because of its association with atypical hemolytic uremic syndrome (aHUS) and other important diseases. Using a combination of biochemical, immunological, nuclear magnetic resonance, and computational approaches, we characterized a series of FHR-1 mutants (including 2 associated with aHUS) and unraveled the molecular bases of the so-called deregulation activity of FHR-1. In contrast with FH, FHR-1 lacks the capacity to bind sialic acids, which prevents C3b-binding competition between FH and FHR-1 in host-cell surfaces. aHUS-associated FHR-1 mutants are pathogenic because they have acquired the capacity to bind sialic acids, which increases FHR-1 avidity for surface-bound C3-activated fragments and results in C3b-binding competition with FH. FHR-1 binds to native C3, in addition to C3b, iC3b, and C3dg. This unexpected finding suggests that the mechanism by which surface-bound FHR-1 promotes complement activation is the attraction of native C3 to the cell surface. Although C3b-binding competition with FH is limited to aHUS-associated mutants, all surface-bound FHR-1 promotes complement activation, which is delimited by the FHR-1/FH activity ratio. Our data indicate that FHR-1 deregulation activity is important to sustain complement activation and C3 deposition at complement-activating surfaces. They also support that abnormally elevated FHR-1/FH activity ratios would perpetuate pathological complement dysregulation at complement-activating surfaces, which may explain the association of FHR-1 quantitative variations with diseases.
Introduction
Factor H (FH)–related protein 1 (FHR-1) originates from a duplication of the gene encoding complement FH. FHR-1 lacks the complement-regulatory domains of FH but presents a highly conserved FH-like C-terminal surface-recognition domain (Figure 1A).1 In FH, this surface-recognition domain includes separate binding sites for the thioester-containing domain (TED) of C3 (closely corresponding to C3d) and sialic acid glycans.2 Crystallographic and nuclear magnetic resonance (NMR) data have provided detailed structural information on these interactions.2-5 The interaction between the C-terminal region of FHR-1 and C3d is much less documented, and whether FHR-1 carries a functional binding site for sialic acid glycans is unknown.5 An additional distinction between FH and FHR-1 is that FHR-1 circulates in plasma as homodimers and heterodimers with FHR-2 as a result of an N-terminal dimerization domain shared by FHR-1, FHR-2, and FHR-5.6,7 It has also been reported that FHR-1 blocks activation of C5 and terminal complex formation.8

Complement deregulation and C3-binding activities of recombinant FHR-1 proteins. (A) Alignment of FH and FHR-1. Numbers in between structures indicate percentages of homology. Proposed FHR-1 dimerization topology is shown. (B) Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis of the recombinant FHR-1 proteins purified from the culture supernatants. (C) FH deregulation assay in sheep erythrocytes (ShEs). Approximately 30% of ShEs are lysed when they are exposed to 20% of a human serum that has been depleted of 75% of the FH. Adding increasing amounts of purified wild-type FHR-1 (FHR-1WT) or FHR-1A296V does not significantly increase this percentage of lysis. This is in contrast with the dose-response hemolysis of ShEs that results from the addition of FHR-1L290S,A296V, FHR-1L290V, and FHR-1L290S. Data are means ± standard deviations of triplicates. (D) Binding to immobilized C3b of the different FHR-1 mutants is compared with that of FHR-1WT using a plate assay. Although FHR-1L290V and FHR-1A296V bind similarly to FHR-1WT (1 ± 0.11 and 0.9 ± 0.4 times, respectively), binding of FHR-1L290S and FHR-1L290S,A296V to C3b was estimated in this assay to be 3.65 ± 0.4 and 4.5 ± 1.56 times lower, respectively, than that of FHR-1WT. (E) FHR-1WT binds similarly to immobilized native C3 (nC3), C3b, iC3b, and C3dg. The same is also true for mutant FHR-1L290S,A296V and all other FHR-1 mutants (data not shown). (F) Binding of FHR-1WT and FH at 300 nM to immobilized C3b and nC3. Only FHR-1 binds to immobilized nC3, indicating that our nC3 preparations were not contaminated with significant amounts of C3(H2O) forms. OD, optical density.
Complement deregulation and C3-binding activities of recombinant FHR-1 proteins. (A) Alignment of FH and FHR-1. Numbers in between structures indicate percentages of homology. Proposed FHR-1 dimerization topology is shown. (B) Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis of the recombinant FHR-1 proteins purified from the culture supernatants. (C) FH deregulation assay in sheep erythrocytes (ShEs). Approximately 30% of ShEs are lysed when they are exposed to 20% of a human serum that has been depleted of 75% of the FH. Adding increasing amounts of purified wild-type FHR-1 (FHR-1WT) or FHR-1A296V does not significantly increase this percentage of lysis. This is in contrast with the dose-response hemolysis of ShEs that results from the addition of FHR-1L290S,A296V, FHR-1L290V, and FHR-1L290S. Data are means ± standard deviations of triplicates. (D) Binding to immobilized C3b of the different FHR-1 mutants is compared with that of FHR-1WT using a plate assay. Although FHR-1L290V and FHR-1A296V bind similarly to FHR-1WT (1 ± 0.11 and 0.9 ± 0.4 times, respectively), binding of FHR-1L290S and FHR-1L290S,A296V to C3b was estimated in this assay to be 3.65 ± 0.4 and 4.5 ± 1.56 times lower, respectively, than that of FHR-1WT. (E) FHR-1WT binds similarly to immobilized native C3 (nC3), C3b, iC3b, and C3dg. The same is also true for mutant FHR-1L290S,A296V and all other FHR-1 mutants (data not shown). (F) Binding of FHR-1WT and FH at 300 nM to immobilized C3b and nC3. Only FHR-1 binds to immobilized nC3, indicating that our nC3 preparations were not contaminated with significant amounts of C3(H2O) forms. OD, optical density.
FHR-1 is associated with various diseases, illustrating remarkable genotype-phenotype correlations. Mutations in the FHR-1 C-terminal domain are prototypical of atypical hemolytic uremic syndrome (aHUS),9,10 and duplications of the N-terminal dimerization domain predispose to C3 glomerulopathy (C3G)6,7 ; FHR-1 deficiency, combined with that of FHR-3 (delCFHR3-CFHR1) is associated with protection from immunoglobulin A nephropathy (IgAN)11 and age-related macular degeneration (AMD)12 and increased risk of systemic lupus erythematosus.13 On the basis of functional analyses of aHUS- and C3G-associated FHR-1 variants, it has been postulated that FHR-1 antagonizes FH regulation by competing with FH in binding to C3b and promoting complement activation.14-16 This FH-antagonistic role of FHR-1 is referred to as complement deregulation activity. A relevant question regarding this deregulation activity of FHR-1 is how its potential harm to host tissues is circumvented in normal conditions or exacerbated in FHR-1 pathogenic variants.
To gain insight into the surface recognition of FHR-1 and how surface-bound FHR-1 promotes complement activation, we generated a series of mutants altering the amino acid positions in the C-terminus of FHR-1 that are different in FH, including 2 mutations associated with aHUS. Using a combination of biochemical, immunological, NMR, and computational approaches, we analyzed the binding of these FHR-1 mutants to C3 molecules and sialic acid glycans. We show how these binding activities determine FHR-1 complement deregulation capacity. Our data reveal the physiological context of the functional competition between FHR-1 and FH and explain the pathogenicity of the FHR-1 variants associated with aHUS and other diseases.
Materials and methods
This study was approved by the ethics committee of the Spanish Consejo Superior de Investigaciones Científicas (reference #SAF2015-66287-R), and all patients included in this study provided written consent.
Generation and purification of recombinant FHR-1 proteins
FHR-1 complementary DNA was purchased from Tebu-Bio Innovative Laboratory Services and Reagents (EX-Z0243-M02), and mutagenesis was performed using the Q5 Site-Directed Mutagenesis Kit (E0552S; New England BioLabs). Mutated FHR-1 complementary DNA was cloned in the pCMV mammalian expression vector and purified by NucleoBond Xtra Maxi (740414.10; Macherey Nagel). Mammalian Hek293 freestyle suspension cells (K900001; Fisher Scientific) were transfected following the manufacturer’s instructions and incubated at 37°C with 5% carbon dioxide at 90-rpm agitation. Culture supernatants were collected after 5 days, and FHR-1 proteins were purified and stored as described for the plasma-derived FHR-1 proteins (supplemental Materials and methods, available on the Blood Web site).
FHR-1 functional assays
The capacity of FHR-1WT and FHR-1 mutants to deregulate FH was assessed using an FH-dependent sheep hemolytic assay. ShEs (Durviz) in AP buffer (2.5 mM of barbital, 1.5 mM of barbital sodium, and 144 mM of sodium chloride [pH, 7.4]) with 5 mM of magnesium chloride and 8 mM of EGTA were incubated with 20% normal human serum (NHS), depleted of 75% of the FH, and increasing amounts of the purified FHR-1 proteins. The reaction was stopped with AP buffer containing 20 mM of EDTA. After centrifugation, supernatants were read at 414 nm, and lysis was plotted vs FHR-1 concentration.
C3-binding assays
Binding of FHR-1 proteins to immobilized nC3, C3b, iC3b, and C3dg was measured using an in-house plate assay. Briefly, 96-well plates were coated with Sim27, a mouse monoclonal antibody (mAb) recognizing an epitope in nC3, C3b, iC3b, and C3dg that does not interfere in the interaction with FHR-1. After blocking the plates with Tris-Tween (50 mM of Tris [pH, 7.4] and 150 mM of sodium chloride with 0.2% Tween20) containing 1% bovine serum albumin (BSA) for 1 hour at room temperature (RT), 100 μL of each of the C3 molecules at saturating concentrations in blocking buffer was added to the wells, and the plates were incubated for 1 hour at RT. After extensive washes, the different FHR-1 proteins were added at different concentrations, and the plates were incubated again for 1 hour at RT. Bound FHR-1 was detected using biotinylated 2C6 (anti-human FHR-1) mAb followed by HRP-conjugated streptoavidin. After incubation with TMB substrate, 0.1 M of sulfuric acid was added to stop the reaction. Absorbance was measured at 450 nm. All samples were tested in triplicate in at least 2 different experiments.
Binding of FHR-1 proteins to cryostat kidney sections
Kidneys were extracted from mice, washed in phosphate-buffered saline (PBS), placed in molds filled with OCT (4583; Tissue-Teck) at 4°C, and immediately immersed in isopentane cooled at −60°C with dry ice. After 5 minutes, molds were transferred to a −80°C freezer. Using Leica CM 1800 cryostat equipment, 5-μm cryostat sections were cut at −18°C and used immediately. For fixing, sections were rinsed in PBS at RT, immersed for 10 minutes in acetone at −20°C, and then extensively washed in PBS at RT. For desialylation, fixed sections were incubated with 50 μL of Clostridium perfringens neuraminidase (Sigma) at 10 U/mL for 90 minutes at 37°C in a humid chamber and then washed extensively in PBS at RT. Sections were incubated with 50 μL of different concentrations of the FHR-1 proteins in PBS (1% BSA) at RT for 40 minutes. Endogenous biotin was blocked using the biotin-blocking system from DAKO (X0590). Biotinylated 2C6 was added at 5 μg/mL in PBS (1% BSA) and incubated at RT for 40 minutes. Sections were finally incubated with streptoavidin Alexa 488 (S32354; Life Technologies), diluted at 1:1000 in PBS (1% BSA) for 30 minutes at RT, incubated with 4′,6-diamidino-2-phenylindole for 2 minutes, and mounted with Moviol. Image analysis was performed with ImageJ software.
Activation of human complement by cryostat kidney sections from Cfh−/−;Cfhr−/− mice
Equal amounts of sera from an FH402H homozygote and FH402H/Y heterozygote were mixed and completely depleted from FHR-1, FHR-2, and FHR-5 by passing the mixture through a sepharose column coupled to the mAb 2C6. Subsequently, the FHR-depleted serum was depleted of 75% of the FH by removing the FH-402H allele using the mAb Mib-7 coupled to a sepharose column. We then added to this serum a sufficient amount of FH to prevent any complement activation upon incubation with cryostat sections from C57bl/6 mouse kidneys and called this serum NHSΔFHR. In a prototypical experiment, cryostat sections from Cfh−/−;Cfhr−/− mouse kidneys were washed in PBS (1% BSA) and then incubated for 30 minutes at 37°C with 10% NHSΔFHR in EGTA-veronal buffer with or without adding FHR-1. After blocking the endogenous biotin, biotinylated 12.17 (anti–human C3 [hC3] mAb that does not recognize mouse C3) was added at 5 μg/mL in PBS (1% BSA) and incubated at RT for 40 minutes. Sections were finally incubated with streptoavidin Alexa 488 (S32354; Life Technologies), diluted at 1:1000 in PBS (1% BSA) for 30 minutes at RT, incubated with 4′,6-diamidino-2-phenylindole for 2 minutes, and mounted with Moviol. Image analysis was performed with ImageJ software.
Statistical analyses
Statistical analyses were performed with SPSS software (version 21). To compare native and mutant FHR-1 functional activities, we used the Student t test for independent samples. P < .05 was considered significant.
NMR spectroscopy (saturation transfer difference NMR [STD-NMR]), molecular dynamic (MD) simulation, and docking calculation are extensively described in the supplemental Materials and methods.
Results
Complement deregulation capacity of recombinant FHR-1 proteins
We have shown that FHR-1L290S,A296V, which carries the C-terminus of FH, deregulates complement in an ShE hemolysis assay and is strongly associated with aHUS.9,10 In addition, a comprehensive genetic analysis of FHR-1 in our large aHUS cohort (n = 640) identified 1 additional FHR-1 genetic variant (FHR-1L290V) that also strongly deregulated complement in the ShE hemolysis assays (supplemental Text). FHR-1L290S,A296V and FHR-1L290V are variants of clinical importance. They explain 1% to 2% of all aHUS cases in our cohort and represent 3% to 4% of all patients carrying complement pathogenic variants. In addition, they are associated with a very poor aHUS prognosis. We generated recombinant proteins corresponding to FHR-1L290S,A296V and FHR-1L290V, single mutants FHR-1L290S and FHR-1A296V, and FHR-1WT (Figure 1B). Analysis of their complement deregulation activity shows that FHR-1L290S,A296V presents the strongest deregulation capacity, followed by FHR-1L290V and FHR-1L290S, whereas FHR-1A296V and FHR-1WT have no deregulation capacity (Figure 1C).
Binding of FHR-1 to nC3 and C3-activated fragments
Unexpectedly, when we tested the binding of FHR-1WT and the FHR-1 mutants to immobilized C3 molecules, we found that all FHR-1 proteins bound nC3 in addition to C3b, iC3b, and C3dg (Figure 1D-E; supplemental Figure 1). We ruled out that our nC3 preparations were contaminated by C3b or C3(H2O) molecules by showing lack of binding of FH to immobilized nC3 (Figure 1F). Furthermore, we failed to detect significant amounts of these molecules in our nC3 preparations upon activation with FB and FD or digestion with FH and FI (data not shown). To provide evidence that the interaction between FHR-1 and nC3 was not an artifact of immobilizing nC3, we confirmed that fluid-phase nC3 bound to immobilized FHR-1 in a dose-dependent manner (supplemental Figure 2).
We found that each of the FHR-1 proteins bound similarly to C3b, iC3b, C3dg, and nC3 (Figure 1E), indicating that FHR-1 interacts with identical binding sites in the 4 C3-related molecules. However, FHR-1L290S,A296V and FHR-1L290S showed decreased binding for the 4 C3-related molecules compared with FHR-1WT, FHR-1L290V, and FHR-1A296V (Figure 1D), which is consistent with previous studies showing that the C-terminus of FHWT has less affinity for C3b than the FHS1191L,V1197A mutant.17 Importantly, these differences in the affinity for the C3-related molecules between the FHR-1 proteins do not correlate with their deregulation activities, indicating that other factors also contribute.
Structural insights into the binding of FHR-1 to the C3-TED domain
Superimposition of the x-ray crystallographic structures of nC3, C3b, and C3dg (C3d; (Protein Data Bank identifiers 2A73, 2I07, and 5NBQ, respectively) showed that the FHR-1–binding site in the TED domain was accessible in all structures (Figure 2A). To gain a 3-dimensional perspective of FHR-1 interactions with the C3 molecules, C3d was selected as the reference protein for docking calculations, and the C-terminal structures of the FHR-1WT and FHR-1 mutants were optimized by MD simulations. Three-dimensional models were built when required (see supplemental Materials and methods). Interestingly, the hypervariable loop in short consensus repeat 5 (SCR5) presented high flexibility (Figures 2B-C). This may justify the capacity of the different FHR-1 proteins to bind to the TED domain, despite the significant structural changes introduced in SCR5 by the amino acid substitutions at positions 290 and 296. The protein-protein docking calculations led to possible binding poses of all the FHR-1 proteins toward C3d at the same binding interface observed in the FH/C3d complex (ie, regions comprising residues 210-226 and 236-241 in SCR4 and residue 289 in SCR5). Other docked poses were obtained at other regions of C3d (data not shown), with FHR-1 always interacting through the same face.
![Structural aspects of FHR-1 interaction with C3 TED and sialic acids. (A) Superimposition of C3 (cyan; Protein Data Bank [PDB] identifier 2A73; TED domain is shown in dark blue) and FHR-1 (yellow)/C3d (pink) complex from MD simulation (100 ns). Key residues are shown in sticks. (B) Root-mean-square deviation (RMSD; calculated with backbone heavy atoms) of FHR-1WT (red) and FHR-1L290S,A296V (black) during 500 ns of MD simulation. (C) Root-mean-square fluctuation (RMSF; calculated by residue) of FHR-1WT (red) and FHR-1L290S,A296V (black) during 500 ns of MD simulation. (D) RMSD (calculated with backbone heavy atoms) of FHR-1/C3dg complex during 500 ns of MD simulation. (E) Electrostatic potential surface of FHR-1L290S,A296V (left) and FHR-1WT (right). The sialic acid–binding sites in SCR-5 of FHR-1L290S,A296V and FHR-1WT are framed by squares that are enlarged below. (F) FH/3′-SL complex (cyan/orange) from docking calculations superimposed to 3′-SL (magenta) from x-ray structure (PDB identifier 4ONT). Key residues are shown in sticks. RMSD of the FH/3′-SL complex (calculated with heavy atoms for protein and with carbons for 3′-SL) from the MD simulation is also depicted (complex in black; 3′-SL in red).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/25/10.1182_blood.2020010069/1/m_bloodbld2020010069f2.png?Expires=1763487319&Signature=PHc6~o91G-ofFYS6izpHWtB7fpB4addnEMvkasUdj7syUN0APY5CiFDyqM7AcozEvQnahBXsXXFbLPUEWjJzrBwkJP37JABUKRIiykZyCM6CqydiTrbAtYcf6kwhzWJauPfFVuCvM92gsCNYffcTEx3dDZInnnHizZQKdUAzbZ~Vc81wUpFBZ~BvpUdTASqLdsvLS8K-UXdA~bCYJyHb4j2N58nCMLulhCI8fSsqTgYPvL9g6QMF2a8y3d7YqTvqGfzbINK6b1x5QTWswHazTR4QR3KQJCz968rXG8txAQAPxyXAvsU57EUJ55G~Mu~4qHl9FUPJxqZVXfuJWvGysw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structural aspects of FHR-1 interaction with C3 TED and sialic acids. (A) Superimposition of C3 (cyan; Protein Data Bank [PDB] identifier 2A73; TED domain is shown in dark blue) and FHR-1 (yellow)/C3d (pink) complex from MD simulation (100 ns). Key residues are shown in sticks. (B) Root-mean-square deviation (RMSD; calculated with backbone heavy atoms) of FHR-1WT (red) and FHR-1L290S,A296V (black) during 500 ns of MD simulation. (C) Root-mean-square fluctuation (RMSF; calculated by residue) of FHR-1WT (red) and FHR-1L290S,A296V (black) during 500 ns of MD simulation. (D) RMSD (calculated with backbone heavy atoms) of FHR-1/C3dg complex during 500 ns of MD simulation. (E) Electrostatic potential surface of FHR-1L290S,A296V (left) and FHR-1WT (right). The sialic acid–binding sites in SCR-5 of FHR-1L290S,A296V and FHR-1WT are framed by squares that are enlarged below. (F) FH/3′-SL complex (cyan/orange) from docking calculations superimposed to 3′-SL (magenta) from x-ray structure (PDB identifier 4ONT). Key residues are shown in sticks. RMSD of the FH/3′-SL complex (calculated with heavy atoms for protein and with carbons for 3′-SL) from the MD simulation is also depicted (complex in black; 3′-SL in red).
Structural aspects of FHR-1 interaction with C3 TED and sialic acids. (A) Superimposition of C3 (cyan; Protein Data Bank [PDB] identifier 2A73; TED domain is shown in dark blue) and FHR-1 (yellow)/C3d (pink) complex from MD simulation (100 ns). Key residues are shown in sticks. (B) Root-mean-square deviation (RMSD; calculated with backbone heavy atoms) of FHR-1WT (red) and FHR-1L290S,A296V (black) during 500 ns of MD simulation. (C) Root-mean-square fluctuation (RMSF; calculated by residue) of FHR-1WT (red) and FHR-1L290S,A296V (black) during 500 ns of MD simulation. (D) RMSD (calculated with backbone heavy atoms) of FHR-1/C3dg complex during 500 ns of MD simulation. (E) Electrostatic potential surface of FHR-1L290S,A296V (left) and FHR-1WT (right). The sialic acid–binding sites in SCR-5 of FHR-1L290S,A296V and FHR-1WT are framed by squares that are enlarged below. (F) FH/3′-SL complex (cyan/orange) from docking calculations superimposed to 3′-SL (magenta) from x-ray structure (PDB identifier 4ONT). Key residues are shown in sticks. RMSD of the FH/3′-SL complex (calculated with heavy atoms for protein and with carbons for 3′-SL) from the MD simulation is also depicted (complex in black; 3′-SL in red).
MD simulations showed high stability at the protein-protein interface in the FHR-1/C3d complexes (Figure 2D). Main FHR-1/C3d interactions at the protein-protein interface involved aliphatic moieties of the side chains from lipophilic, aromatic, and cationic residues: P213-N1163, G217-L1109, D128-K1105, F222-Q1098, F222-Q1161, P223-Q1161, Q236-L1109, N239-K1113, L240-K1113, L240-P1114, Y241-P1114, P265-P1114, and Y286-P1114. Also, H bonds were found between D218-N1163, D218-S1164, Q238-Q111, Q238-I1108, N239-L1109, N239-E1110, and Y289-D1115 and a π-cation interaction between the side chains of Y289 and K1113. Because our computational results show that all FHR-1/C3d interactions are stable, we propose these models as likely complexes accounting for the binding of the FHR-1 proteins to nC3 and its activated fragments.
Binding of FHR-1WT and FHR-1 mutants to C3-opsonized physiological surfaces
The differences in binding C3-related molecules between the FHR-1 proteins do not justify their distinct complement deregulation activities in the ShE assays (Figure 1C). We therefore moved our binding experiments to cryostat sections from Cfh−/−18 and Cfh−/−;Cfhr−/− (Matthew Pickering, unpublished data) mouse glomeruli. The rationale for this was that glomeruli from these mice are C3-opsonized physiological surfaces that present both C3-activated fragments and physiological polyanions (Figure 3A). In these experiments, FHR-1L290S,A296V, FHR-1L290V, and FHR-1L290S bound much better to C3-opsonized mouse glomeruli than FHR-1WT and FHR-1A296V (Figure 3B), and their relative binding correlated well with their complement deregulation activities in the ShE assays. The interactions with the mouse glomeruli cannot be explained considering only the binding of the FHR-1 proteins to the C3-activated molecules; this suggests that the strong binding of FHR-1L290S,A296V, FHR-1L290V, and FHR-1L290S mutants involves other surface ligands. Notably, none of the FHR-1 recombinant proteins bound to mouse glomeruli lacking C3 deposits (supplemental Figure 3), and binding of the different FHR-1 proteins to C3-opsonized mouse glomeruli was not altered by the presence of a 4-M excess of FH (supplemental Figure 4).
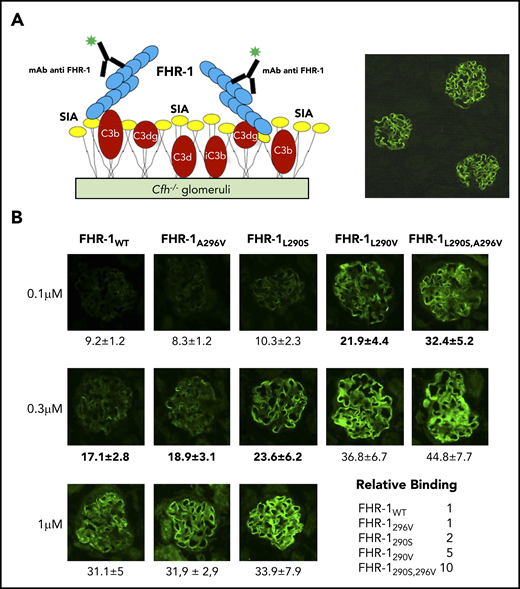
Binding of FHR-1 to glomeruli from Cfh−/− mice. (A) Illustration of the rationale for the experiments using cryostat sections of Cfh−/− mouse kidneys. Photomicrograph shows staining with a biotinylated anti–mouse C3 antibody. (B) Three different concentrations of FHR-1 proteins were added to cryostat kidney sections of Cfh−/− mice. Bound FHR-1 was detected with an in-house biotinylated anti–FHR-1 mAb (2C6). All FHR-1 proteins bound exclusively to the glomerulus. Fluorescence intensity (FI) is expressed in arbitrary units. Data are means ± standard deviations of a minimum of 20 glomeruli. FI values obtained from FHR-1WT were used to make a reference curve. FI values of all FHR-1 mutants were interpolated in this curve to calculate the relative binding between the FHR-1 proteins. SIA, sialic acid. All photomicrographs were taken at ×40 magnification. Average diameter of mouse glomeruli is 70 μm.
Binding of FHR-1 to glomeruli from Cfh−/− mice. (A) Illustration of the rationale for the experiments using cryostat sections of Cfh−/− mouse kidneys. Photomicrograph shows staining with a biotinylated anti–mouse C3 antibody. (B) Three different concentrations of FHR-1 proteins were added to cryostat kidney sections of Cfh−/− mice. Bound FHR-1 was detected with an in-house biotinylated anti–FHR-1 mAb (2C6). All FHR-1 proteins bound exclusively to the glomerulus. Fluorescence intensity (FI) is expressed in arbitrary units. Data are means ± standard deviations of a minimum of 20 glomeruli. FI values obtained from FHR-1WT were used to make a reference curve. FI values of all FHR-1 mutants were interpolated in this curve to calculate the relative binding between the FHR-1 proteins. SIA, sialic acid. All photomicrographs were taken at ×40 magnification. Average diameter of mouse glomeruli is 70 μm.
FHR-1WT does not bind to sialic acid glycans
Removal of the sialic acid glycans from the C3-opsonized mouse glomeruli using C perfringens neuraminidase did not affect binding of FHR-1WT or FHR-1A296V but greatly reduced the binding of FHR-1L290S,A296V, FHR-1L290V, and FHR-1L290S mutants to this surface (Figure 4). All FHR-1 proteins bound to neuraminidase-treated mouse glomeruli, but FHR-1L290S,A296V and FHR-1L290S showed weaker interaction, which reflects their slightly reduced binding to C3-activated fragments compared with FHR-1WT, FHR-1L290V, and FHR-1A296V (Figure 1D). These experiments suggest that sialic acid glycans mediate the strong binding of the mutants FHR-1290V, FHR-1290S, and FHR-1290S,296V to C3-opsonized mouse glomeruli. To provide formal proof that FHR-1L290S, FHR-1L290V, and FHR-1L290S,A296V bind sialic acid glycans and FHR-1WT and FHR-1A296V do not, we performed STD-NMR19 experiments using 2,3-sialyllactose (3′-SL) and 2,6-sialyllactose (6′-SL) as analogs of α2-3– and α2-6–linked sialic acid glycans, respectively (Figure 5A). To validate our STD-NMR experiments, we first replicated the experiments reported by Blaum et al2 showing that plasma purified FHWT binds 3′-SL but not 6′-SL (supplemental Figure 5). Then, we analyzed FHR-1WT, and consistently with the data from the C3-opsonized mouse glomeruli experiments, we found that FHR-1WT did not bind to 3′-SL or 6′-SL. Importantly, FHR-1L290S,A296V, which carries the C-terminus of FH, bound 3′-SL but not 6′-SL (Figure 5B; supplemental Figure 6). We also included in these experiments the mutant FHS1191L,V1197A, purified from the plasma of an aHUS patient, in which the C-terminus of FH was substituted with the C-terminus of FHR-1. The results of this experiment were in agreement with the data obtained from FHR-1WT showing that the FHS1191L,V1197A mutant did not bind to 3′-SL or 6′-SL (supplemental Figures 5 and 6). Our STD-NMR experiments also showed that FHR-1L290S bound to 3′-SL and that FHR-1A296V did not bind to 3′-SL or 6′-SL (Figure 5B; supplemental Figure 6). Interestingly, STD-NMR data showed that mutant FHR-1L290V bound 6′-SL instead of 3′-SL (Figure 5C). In all cases where interaction was observed, the methyl of the 5-NHAc group of the sialic acid moiety received the highest saturation, indicating close contact with the protein.

Decreased binding of aHUS-associated FHR-1 mutants to desialylated Cfh−/− mouse glomeruli. Optimal concentrations of FHR-1WT (1 μM), FHR-1L290V (0.3 μM), FHR-1296A (1 μM), FHR-1L290S (0.3 μM), and FHR-1L290S,A296V (0.3 μM) in PBS/BSA were added to normal and neuraminidase (NA)-treated cryostat sections of Cfh−/− mouse kidneys and incubated at 37°C for 30 minutes. Bound FHR-1 was detected with an anti–FHR-1 mAb (2C6). Images of representative glomeruli are shown to illustrate the decreased binding to desialylated glomeruli of the aHUS-associated FHR-1 mutants compared with the binding to untreated glomeruli. Loss of binding for the different FHR-1 proteins was calculated basically as described in Figure 3. All FHR-1 proteins bound similarly to desialylated sections of Cfh−/− mouse kidneys when they were used at the same concentration (data not shown). All photomicrographs were taken at ×40 magnification.
Decreased binding of aHUS-associated FHR-1 mutants to desialylated Cfh−/− mouse glomeruli. Optimal concentrations of FHR-1WT (1 μM), FHR-1L290V (0.3 μM), FHR-1296A (1 μM), FHR-1L290S (0.3 μM), and FHR-1L290S,A296V (0.3 μM) in PBS/BSA were added to normal and neuraminidase (NA)-treated cryostat sections of Cfh−/− mouse kidneys and incubated at 37°C for 30 minutes. Bound FHR-1 was detected with an anti–FHR-1 mAb (2C6). Images of representative glomeruli are shown to illustrate the decreased binding to desialylated glomeruli of the aHUS-associated FHR-1 mutants compared with the binding to untreated glomeruli. Loss of binding for the different FHR-1 proteins was calculated basically as described in Figure 3. All FHR-1 proteins bound similarly to desialylated sections of Cfh−/− mouse kidneys when they were used at the same concentration (data not shown). All photomicrographs were taken at ×40 magnification.
![NMR data for the interaction of FHR-1 proteins with 3′-SL and 6′-SL. (A) Structures of 3′-SL (α-Neu5Ac-[2-3]-β-D-Gal-[1-4]-D-Glc) and 6′-SL (α-Neu5Ac-[2-6]-β-D-Gal-[1-4]-D-Glc). (B) STD-NMR spectra with 3′-SL. Lower panel shows reference spectra of 3′-SL corresponding to the off-resonance spectra acquired in presence of FHR-1WT. Above this are panels showing the STD spectra in presence of FHR-1WT at 9 µM and FHR-1L290S, A296V at 16 µM; the acetate of the 5-NAc group of the sialic residue received the highest saturation. In both cases, 3′-SL concentration was adjusted to 50:1 ligand/protein ratio. (C) STD-NMR spectra of FHR-1L290V mutant with 3′-SL and 6′-SL. From bottom to top, panels show reference spectra of 3′-SL corresponding to the off-resonance spectra in presence of FHR-1L290V, STD-NMR spectra of 3′-SL in presence of FHR-1L290V at 15 µM, reference spectra of 6′-SL corresponding to the off-resonance spectra in presence of FHR-1L290V, and STD-NMR spectra of 6′-SL in presence of FHR-1L290V at 16 µM; the acetate of the 5-NAc group of the sialic residue received the highest saturation. In both cases, 3′-SL and 6′-SL concentrations were adjusted to 50:1 ligand/protein ratio. All STD-NMR spectra were acquired with 3-s saturation on −0.14 ppm. Background signal of protein was subtracted in each case from a spectrum of the protein acquired under the same conditions but in absence of ligand. Signals of selected protons are labeled.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/25/10.1182_blood.2020010069/1/m_bloodbld2020010069f5.png?Expires=1763487319&Signature=HZ~kdE500CfWVhBfpzZDd0ztIeIeY9IIzg2aAyMSwDGh-H0HFeMu44l3JTRK-BpvcYBMjC~D9E~ay-wNUuhIclsWRxVu8-PRtDjlOZaR8gkMgyns75NMAgrJ~azgCJwwBudbDxW-PcJVU~-OO5G7kciL8JnbndmGkSUM1B~C2bwRsGzbNCPCgBjpT9uadurE9clOFJcY-DrVVUgEU0Z6~V6-8jzD1upOD2nGcOO62w9~tTxSnylBHMLOyCAgKFOL8QUXhMPF2fTr-ZYd2Wu5NBSqqqx9UCGQv0I0-HMmdlVaKNBWCad~PR6UOOPYZRwJIqzbebaQ0al~bxdmQTquGw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
NMR data for the interaction of FHR-1 proteins with 3′-SL and 6′-SL. (A) Structures of 3′-SL (α-Neu5Ac-[2-3]-β-D-Gal-[1-4]-D-Glc) and 6′-SL (α-Neu5Ac-[2-6]-β-D-Gal-[1-4]-D-Glc). (B) STD-NMR spectra with 3′-SL. Lower panel shows reference spectra of 3′-SL corresponding to the off-resonance spectra acquired in presence of FHR-1WT. Above this are panels showing the STD spectra in presence of FHR-1WT at 9 µM and FHR-1L290S, A296V at 16 µM; the acetate of the 5-NAc group of the sialic residue received the highest saturation. In both cases, 3′-SL concentration was adjusted to 50:1 ligand/protein ratio. (C) STD-NMR spectra of FHR-1L290V mutant with 3′-SL and 6′-SL. From bottom to top, panels show reference spectra of 3′-SL corresponding to the off-resonance spectra in presence of FHR-1L290V, STD-NMR spectra of 3′-SL in presence of FHR-1L290V at 15 µM, reference spectra of 6′-SL corresponding to the off-resonance spectra in presence of FHR-1L290V, and STD-NMR spectra of 6′-SL in presence of FHR-1L290V at 16 µM; the acetate of the 5-NAc group of the sialic residue received the highest saturation. In both cases, 3′-SL and 6′-SL concentrations were adjusted to 50:1 ligand/protein ratio. All STD-NMR spectra were acquired with 3-s saturation on −0.14 ppm. Background signal of protein was subtracted in each case from a spectrum of the protein acquired under the same conditions but in absence of ligand. Signals of selected protons are labeled.
NMR data for the interaction of FHR-1 proteins with 3′-SL and 6′-SL. (A) Structures of 3′-SL (α-Neu5Ac-[2-3]-β-D-Gal-[1-4]-D-Glc) and 6′-SL (α-Neu5Ac-[2-6]-β-D-Gal-[1-4]-D-Glc). (B) STD-NMR spectra with 3′-SL. Lower panel shows reference spectra of 3′-SL corresponding to the off-resonance spectra acquired in presence of FHR-1WT. Above this are panels showing the STD spectra in presence of FHR-1WT at 9 µM and FHR-1L290S, A296V at 16 µM; the acetate of the 5-NAc group of the sialic residue received the highest saturation. In both cases, 3′-SL concentration was adjusted to 50:1 ligand/protein ratio. (C) STD-NMR spectra of FHR-1L290V mutant with 3′-SL and 6′-SL. From bottom to top, panels show reference spectra of 3′-SL corresponding to the off-resonance spectra in presence of FHR-1L290V, STD-NMR spectra of 3′-SL in presence of FHR-1L290V at 15 µM, reference spectra of 6′-SL corresponding to the off-resonance spectra in presence of FHR-1L290V, and STD-NMR spectra of 6′-SL in presence of FHR-1L290V at 16 µM; the acetate of the 5-NAc group of the sialic residue received the highest saturation. In both cases, 3′-SL and 6′-SL concentrations were adjusted to 50:1 ligand/protein ratio. All STD-NMR spectra were acquired with 3-s saturation on −0.14 ppm. Background signal of protein was subtracted in each case from a spectrum of the protein acquired under the same conditions but in absence of ligand. Signals of selected protons are labeled.
Sialic acid binding site in FHR-1WT and FHR-1 mutants
The binding site in the FH/3′-SL complex (Protein Data Bank identifier 4ONT; identical to the FHR-1L290S,A296V mutant) presented a well-defined cavity to accommodate the glycerol chain in 1 side and the N-acetyl group in the other side (Figure 2E). The electrostatic potential surface (EPS) of this cavity in FH is mainly neutral, with some polar residues at the entrance: the R1215 (R314 in FHR-1) side chain, which interacts with the sialic carboxylate group, and the E1195 (E294 in FHR-1) side chain placed in between the electron-deficient N-acetyl carbonyl carbon and the OH group at position 4 of the sialic acid. Another key residue for ligand recognition in FH is W1183 (W282 in FHR-1), the side chain of which establishes CH-π interactions with CH groups from the glucose and galactose moieties. The interaction between Gal OH-6 and E1198 (E297 in FHR-1) completed the picture of the 3′-SL–binding site in FH (Figure 2F). Molecular docking calculations of 3′-SL and 6′-SL were performed, predicting the x-ray crystallographic pose for 3′-SL, but they failed to provide appropriate binding poses for 6′-SL (Figure 2F).
In FHR-1WT, the sialic acid–binding site was closed, disappearing completely as cavity, and the EPS was predominantly positive because of the location of the K287 side chain (Figure 2E; supplemental Figure 7). The W282 side chain pointed backward to the binding site, losing the capacity to anchor 3′-SL through CH-π interactions. Docking calculations were performed and did not predict suitable binding poses for either 3′-SL or 6′-SL. In FHR-1L290V, the sialic acid cavity was only partially closed (supplemental Figure 7). The negatively charged E294 side chain, also important to anchor the sialyl moiety, was located farther from the binding site because of the change in the orientation of the E294 side chain, whereas the positively charged R314 was located closer to the binding site. The W282 side chain was also pointing backward to the binding site, not allowing the establishment of CH-π interactions with the lactose moiety. In fact, docking calculations of 3′-SL did not result in favorable docked poses, in contrast with 6′-SL, for which suitable docked poses were mainly predicted through interaction with the sialic acid moiety. MD simulations of the FHR-1L290V/6′-SL system confirmed the stability of the complex, in agreement with the experimental observations (supplemental Figure 8). In FHR-1A296V, the sialic acid cavity remained almost completely closed during the MD simulation, and docking calculations did not lead to good predicted binding poses for the ligand (supplemental Figure 7). For FHR-1L290S, a cavity at the sialic acid–binding site was clearly observed, although with a different architecture because of the loop conformation, which pushed W282 backward from and K287 toward the entrance of the sialic-binding site (supplemental Figure 7). Docking calculations of 3′-SL predicted appropriate binding poses similar to those in FH, with the exception of the glucose moiety, which, because W282 was not sufficiently close to interact, pointed toward T283, also located at the loop. These docked poses accounted for the observed weak binding.
Taken together, STD-NMR and computational data provide compelling molecular evidence demonstrating that FHR-1WT does not bind sialic acids and explaining the acquisition of sialic acid–binding capacity by the FHR-1L290S,A296V, FHR-1L290S, and FHR-1L290V mutants.
Surface-bound FHR-1 proteins promote complement activation
To determine whether the surface-bound FHR-1 mutant proteins promote complement activation in a physiological context, kidney cryostat sections from Cfh−/−; Cfhr−/− mice were incubated with human serum–EGTA depleted completely of FHR-1, FHR-2, and FHR-5 (NHSΔFHR) that was reconstituted with the different FHR-1 recombinant proteins. These experiments showed that FHR-1L290S,A296V, FHR-1L290V, and FHR-1L290S bound to the C3-opsonized Cfh−/−;Cfhr−/− mouse glomeruli promoted significantly more activation and deposition of hC3 than FHR-1WT and FHR-1A296V (Figure 6A). The deposition of hC3 in Cfh−/−;Cfhr−/− mouse glomeruli could be prevented by the presence of 20 mM of EDTA, indicating that C3 deposition requires formation of the alternative pathway C3 convertase. Importantly, C3 deposition was also prevented by the addition of a fourfold molar excess of FH with respect to FHR-1 (Figure 6B), but this molar excess of FH did not prevent binding of the FHR-1 proteins to the Cfh−/−;Cfhr−/− mouse glomeruli (Figure 6C).

Surface-bound FHR-1 promotes complement activation. (A) Cryostat kidney sections from Cfh−/−;Cfhr−/− mice were incubated with 10% NHSΔFHR in presence or absence of 0.5 µM of the different FHR-1 proteins. Bound FHR-1 was detected using the mAb 2C6, and deposits of hC3-activated fragments were detected with the mAb 12.17. (B) Cryostat kidney sections from Cfh−/−;Cfhr−/− mice were incubated with 10% NHSΔFHR and 0.25 µM of FHR-1L290S,A296V in the presence or absence of FH (1 µM) or EDTA (20 mM). (C) Increasing concentrations of FHR-1WT or FHR-1L290S,A296V were added to 10% NHSΔFHR and then incubated with cryostat kidney sections from Cfh−/−;Cfhr−/− mice. Fluorescence intensity is expressed in arbitrary units. Data are means ± standard deviations of a minimum of 40 glomeruli. All photomicrographs were taken at ×40 magnification.
Surface-bound FHR-1 promotes complement activation. (A) Cryostat kidney sections from Cfh−/−;Cfhr−/− mice were incubated with 10% NHSΔFHR in presence or absence of 0.5 µM of the different FHR-1 proteins. Bound FHR-1 was detected using the mAb 2C6, and deposits of hC3-activated fragments were detected with the mAb 12.17. (B) Cryostat kidney sections from Cfh−/−;Cfhr−/− mice were incubated with 10% NHSΔFHR and 0.25 µM of FHR-1L290S,A296V in the presence or absence of FH (1 µM) or EDTA (20 mM). (C) Increasing concentrations of FHR-1WT or FHR-1L290S,A296V were added to 10% NHSΔFHR and then incubated with cryostat kidney sections from Cfh−/−;Cfhr−/− mice. Fluorescence intensity is expressed in arbitrary units. Data are means ± standard deviations of a minimum of 40 glomeruli. All photomicrographs were taken at ×40 magnification.
Discussion
We provide compelling evidence indicating that a role of FHR-1 is binding to C3-opsonized surfaces to promote complement activation and further deposition of C3-activated fragments. We also show that the 2 amino acid differences that distinguish the C-terminal region of FHR-1WT from that of FH confer to FHR-1WT distinctive binding characteristics that prevent C3b competition with FH and potential damage to normal host tissues. An unexpected finding of this study is that FHR-1 interacts with nC3 similarly to C3b, iC3b, and C3dg, which suggests that promotion of complement activation by surface-bound FHR-1 may involve attracting nC3 to the surface rather than capturing fluid-phase C3b to organize C3 convertase. Notably, although C3b-binding competition with FH was limited to aHUS-associated mutants, all surface-bound FHR-1 promoted complement activation. This promotion depended on the amount of FHR-1 bound to the surface, and it was regulated by the levels of fluid-phase FH (FHR-1/FH activity ratio). A summary of our understanding of the interaction of FHR-1WT and FHR-1 mutants with normal host cells, altered host surfaces, and pathogens and their consequences in complement regulation is depicted in Figure 7.

Interaction of FHR-1WT and FHR-1 mutants with normal host cells and altered host surfaces and pathogens and their consequences in complement regulation.
Interaction of FHR-1WT and FHR-1 mutants with normal host cells and altered host surfaces and pathogens and their consequences in complement regulation.
Previous analyses of aHUS-associated FHR-1 mutants have suggested that the 2 amino acid differences that distinguish the C-terminal region of FH from that of FHR-1WT are crucial to prevent complement deregulation by FHR-1WT in normal host-cell surfaces.1 Here, we found that these 2 amino acid positions conferred on FH and FHR-1WT distinct interaction with the TED domain of C3 and sialic acid glycans. The arrangement 290Leu-296Ala, which is the situation found in FHR-1WT, resulted in increased interaction with the TED domain in nC3, C3b, iC3b, and C3dg, compared with the sequence arrangement found in FH (290Ser-296Val), and in the loss of sialic acid–binding capacity. These binding differences between FHR-1WT and FH may explain the lack of complement deregulation of FHR-1WT in host-cell surfaces. It is unlikely that FHR-1WT, lacking sialic acid–binding capacity, could efficiently compete with FH in binding to the low-density (physically distant) C3b molecules accidentally deposited on cell surfaces decorated with sialic acid glycans. The simultaneous binding of the N-terminal and C-terminal regions of FH to separate regions in C3b and the concurrent recognition of sialic acid glycans result in a strong interaction of FH with surface-bound C3b that facilitates regulation.3,5 In contrast, FHR-1WT should bind efficiently to the C3-activated fragments (mostly iC3b and C3dg) that are present in high density in C3-opsonized surfaces, whereas FH binds weakly to these ligands.
Experiments using cryostat sections from Cfh−/−;Cfhr−/− mouse kidneys have shown that surface-bound FHR-1 promotes complement activation and increases deposition of C3. In previous reports, we have speculated that surface-bound FHR-1 may work as a platform that promotes complement activation through the recruitment of fluid-phase C3b and the organization of C3 convertase.20 Our finding that FHR-1 binds equally to C3b and nC3 now makes that possibility unlikely. Under normal physiological conditions, the amounts of C3b in plasma that escape inactivation by FH must be very low, and its binding to FHR-1 was easily challenged by the high concentration of nC3 in plasma (supplemental Figure 2B). Our current view is that by interacting with nC3, surface-bound FHR-1 raises the concentration of nC3 toward the proximity of the surface, increasing the spontaneous deposition of C3b-like molecules generated by spontaneous or protease-mediated C3 activation. Under normal circumstances, this activation of C3 and subsequent generation of C3 convertase are likely controlled by the regulatory activity of FH. This may even be beneficial at opsonized surfaces, because a continuous generation of iC3b molecules would facilitate removal by opsonophagocytosis through interactions with the CR3 integrin receptor. However, if this FHR-1-mediated promotion of complement activation were to exceed the regulatory capacity of FH, or the levels of FH were low, it would cause complement dysregulation at those surfaces.
Central to our study is the finding that the 2 amino acid differences between the C-terminus of FH and FHR-1WT eliminate the capacity to bind to sialic acid glycans in FHR-1WT and that this ability is recovered in the aHUS-associated FHR-1 mutants. This explains why the aHUS-associated FHR-1 proteins are pathogenic. Acquisition of sialic acid–binding capacity allows them to compete efficiently in the binding and regulation by FH of surface-bound C3b. They would also be able to bind low-density iC3b and C3dg deposited in host-cell surfaces, promoting complement activation. Other situations in which FHR-1 becomes pathogenic occur in the FHR-1 mutants with duplicated dimerization domains. These mutants organize large multimers with increased avidity for C3-activated fragments and are strongly associated with C3G.7 A distinction between these C3G-associated FHR-1 mutants and those associated with aHUS is that the former, lacking sialic acid–binding capacity, require high-density C3 deposits for binding. This peculiarity likely restricts the pathogenicity of the FHR-1 multimers that characterizes the C3G-associated FHR-1 mutants to the context of C3-opsonized surfaces.
Plasma levels of FHR-1 vary widely in humans as a consequence of genetic and environmental factors, and the same is true for FH.10,14,21-23 These quantitative variations in the context of a functional competition between FH and FHR-1WT at C3-opsonized surfaces explain the association of FHR-1 with increased risk or protection against various diseases. IgAN and AMD are examples in which genetically determined decreased levels of FHR-1 (delCFHR3-CFHR1) are associated with strong protection from development of the disease.11,12 The pathogenesis of IgAN and AMD is currently shaped as a sequence of multiple events ending in the generation of altered surfaces at the glomerular mesangium and the macular neuroretina (ie, by immune complex deposits or MAA modifications), which activate complement and generate deposition of C3-activated fragments. Individuals carrying delCFHR3-CFHR1 have less FHR-1 to bind these C3-opsonized surfaces and promote complement activation and further C3 deposition, which potentiates regulation by FH. In contrast, delCFHR3-CFHR1 has been described as a predisposing factor for systemic lupus erythematosus, a severe disease characterized by the presence of autoantibodies that mediate tissue injury at various organs.13 Decreased levels of FHR-1 could reduce C3 deposition and opsonophagocytic removal of apoptotic cells, which may result in increased levels of pathogenic autoantibodies.
In conclusion, we have revealed why the aHUS-associated FHR-1 mutants are pathogenic. In addition, our data illustrate that the complement deregulation activity of FHR-1 is a 2-step process: the binding of FHR-1 to the cell surface, which is influenced by FHR-1 concentration and its avidity for surface-bound C3-activated fragments, and the promotion of complement activation by surface-bound FHR-1, which is mediated by the interaction with nC3 and modulated by the level and activity of FH. Importantly, with the exception of the aHUS-associated FHR-1 mutants, FHR-1 binds weakly to low-density C3b deposited on host surfaces, which eliminates competition between FHR-1 and FH and prevents potential damage to normal host tissues by FHR-1 deregulation activity. This novel mechanistic understanding of the deregulation activity of FHR-1 as a balance between the activity of surface-bound FHR-1 (promoting complement activation) and that of fluid-phase FH (regulating complement activation) provides a conceptual framework that explains the association of FHR-1 mutants with aHUS and other diseases. Finally, our data suggest that complement opsonization is a context in which FHR-1 likely plays a physiological role.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Matthew Pickering for sending us kidneys from Cfh−/−;Cfhr−/− mice before reporting the description of these mice. The authors acknowledge the contribution of Hugo Yebenes in the very early stages of this work, suggesting that FHR-1 interacts with native C3. The authors also thank Josue Gutierrez-Tenorio for helping with FHR-1WT cloning and providing some FHR-1WT protein at the beginning of the work.
S.R.d.C., F.J.C., and S.M.-S. are supported by Spanish “Ministerio de Economía y Competitividad-FEDER” grants (SAF2015-66287-R and PID2019-104912RB-I00, RTI2018-094751-B-C22, and CTQ2017-88353-R, respectively), and Autonomous Region of Madrid grant S2017/BMD-3673. E.G.d.J.;is supported by Spanish “Ministerio de Ciencia, Innovación y Universidades-FEDER grants RYC-2013-13395 and RTI2018-095955-B-100.
Authorship
Contribution: E.G.d.J., S.M.-S., F.J.C., and S.R.d.C. designed the experiments; H.M.M., M.S., A.P., and E.G.-R. performed the research and collected the data; L.J.L. performed the animal work; C.F. provided the clinical data; H.M.M., M.S., A.P., E.G.-R., S.R.d.C., E.G.d.J., S.M.-S., and F.J.C. analyzed the data; and S.R.d.C., S.M.-S., and F.J.C. wrote the manuscript, which was revised by all other authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Santiago Rodríguez de Córdoba, Centro de Investigaciones Biológicas Margarita Salas, Ramiro de Maeztu 9, 28040 Madrid, Spain; e-mail: srdecordoba@cib.csic.es.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal