

Key Points
The number of rare nonsynonymous VWF variants is significantly associated with VWF:Ag levels, regardless of VWD type.
VWF sequence alone will not reveal the cause of VWD in a majority of patients with higher VWF:Ag levels.

Abstract
Approximately 35% of patients with type 1 von Willebrand disease (VWD) do not have a known pathogenic variant in the von Willebrand factor (VWF) gene. We aimed to understand the impact of VWF coding variants on VWD risk and VWF antigen (VWF:Ag) levels, studying 527 patients with low VWF and VWD and 210 healthy controls. VWF sequencing was performed and VWF:Ag levels assayed. A combined annotation-dependent depletion (CADD) score >20 was used as a predicted pathogenicity measure. The number of rare nonsynonymous VWF variants significantly predicted VWF:Ag levels (P = 1.62 × 10−21). There was an association between average number of rare nonsynonymous VWF variants with VWD type 1 (P = 2.4 × 10−13) and low VWF (P = 1.6 × 10−27) compared with healthy subjects: type 1 subjects possessed on average >2 times as many rare variants as those with low VWF and 8 times as many as healthy subjects. The number of rare nonsynonymous variants significantly predicts VWF:Ag levels even after controlling for presence of a variant with a CADD score >20 or a known pathogenic variant in VWF (P = 2.7 × 10−14). The number of rare nonsynonymous variants in VWF as well as the presence of a variant with CADD >20 are both significantly associated with VWF levels. The association with rare nonsynonymous variants holds even when controlling for known pathogenic variants, suggesting that additional variants, in VWF or elsewhere, are associated with VWF:Ag levels. Patients with higher VWF:Ag levels with fewer rare nonsynonymous VWF gene variants could benefit from next-generation sequencing to find the cause of their bleeding.
Introduction
von Willebrand disease (VWD) type 1, the most common type of VWD, is characterized by low plasma von Willebrand factor (VWF) levels with normal VWF structure and function, and is primarily transmitted in an autosomal-dominant inheritance pattern.1 It is mainly associated with haploinsufficiency at the VWF locus.2 Patients with VWD type 1 are further subcategorized into a low VWF phenotype or type 1 VWD based on VWF plasma levels of 0.03 to 0.3 U mL−1 and 0.3 to 0.5 U mL−1, respectively,3 although these cutoffs have long been debated.4,5 Type 2 VWD is characterized by functional defects in the VWF molecule, and patients with type 3 VWD have a near complete absence of VWF.6 Thus, it is a phenotypically heterogeneous disease.
Family studies have put the heritability of VWF levels at ∼30%.7 However, twin studies have set estimates of the heritability of VWF plasma levels at as high as 75%,8 with 30% of the genetic variance explained by the effect of ABO blood type and only ∼5% explained by variation in the VWF gene itself.9 In fact, ∼35% of patients with type 1 VWD do not have a known pathogenic variant in the VWF gene,10 and a previous linkage study found that only 41% of families with VWD type 1 had linkage to the VWF locus.11 This figure is even lower in patients with low VWF.6
Thus, VWD type 1 and low VWF are likely indicative of complex disorders with multiple genetic risk factors. With VWD, as with many other disorders, the more severe cases, such as type 2 and 3, are enriched for rare and putatively causative mutations in the VWF gene. However, the low VWF and type 1 VWD cases represent a milder form of the disorder that exists on a spectrum of severity, which will necessarily have a more complicated genetic basis that more often involves variants in other genes besides VWF.
As a class, rare variants (≤1% population frequency) constitute the majority of genetic variation and are 4 times more likely to be deleterious.12 Previous studies of VWF genetics have relied largely on genotyping arrays and genome-wide association studies of VWF antigen (VWF:Ag) levels.13-17 These techniques, however, do not reliably detect rare variants, which to date are the class of variants that have explained the largest proportion of VWD cases.
In a number of genetic disorders, it has been shown that patients with multiple rare variants in causative genes have poorer outcomes than those with fewer rare variants in the same genes, in terms of disease onset, severity, and prognosis. This phenomenon has been observed in amyotrophic lateral sclerosis,18 schizophrenia,19 and Charcot-Marie-Tooth.20 Here, we aim to determine whether the number of rare and predicted deleterious nonsynonymous variants in the VWF gene correlates with VWF:Ag levels and VWD type.
Methods
Cohort description
All subjects in the present study were recruited from 35 sites through the Zimmerman Program for the Molecular and Clinical Biology of VWD, or the Zimmerman Program. All recruited patients in the present study were unrelated. The institutional review board of each study center approved this study and all patients and/or parents provided informed consent. A preexisting diagnosis of VWD of any type was required for participation in the study. Here, we use the laboratory phenotypic diagnosis assigned to patients from the central laboratory values obtained at the time of study entry as per the National Heart, Lung, and Blood Institute (NHLBI) guidelines (Table 1). For type 1 subjects, we used VWF:Ag or VWF ristocetin cofactor (VWF:RCo) <30 IU/dL and for low VWF we used VWF:Ag or VWF:RCo 30 to 50 IU/dL at the time of study entry. Those patients with a historical diagnosis of type 1 VWD but normal VWF:Ag levels when tested by the central laboratory were excluded. Controls comprised 210 healthy individuals with a negative bleeding history, with VWF sequence data and laboratory results completed. DNA was obtained from blood samples collected from each subject at the time of enrollment. All patients were assayed in the research laboratory at Versiti Blood Research Institute for VWF:Ag, VWF propeptide, VWF mutant glycoprotein Ib binding, VWF collagen III binding, and VWF collagen IV binding. Bleeding questionnaires were also administered to each subject, and bleeding scores were determined using the International Society on Thrombosis and Haemostasis Bleeding Assessment Tool (ISTH BAT) scoring system (see Flood et al for a full description21 ) (supplemental Table 1, available on the Blood Web site). Sixty-four percent of patients were female, 87% of patients were White, 5% were African American, 2% were Asian, and 6% were unknown. Four patients with type 3 VWD on treatment were removed, as were 3 patients having a diagnosis of unclassified VWD. This left 737 patients in the present analyses.
Sample distribution
| VWD type | N |
|---|---|
| Healthy controls | |
| Total | 210 |
| Low VWF | |
| Total | 169 |
| Type 1 | |
| Type 1 | 78 |
| Type 1 severe | 11 |
| Type 1/HA or HA carrier | 1 |
| Type 1C | 63 |
| Total | 153 |
| Type 2 | |
| Type 2A | 73 |
| Type 2B | 52 |
| Type 2M | 24 |
| Type 2M-C | 2 |
| Type 2N | 9 |
| Type 2N carrier | 1 |
| Type 2N/type 1 | 4 |
| Total | 165 |
| Type 3 | |
| Total | 40 |
| Overall total | 737 |
| VWD type | N |
|---|---|
| Healthy controls | |
| Total | 210 |
| Low VWF | |
| Total | 169 |
| Type 1 | |
| Type 1 | 78 |
| Type 1 severe | 11 |
| Type 1/HA or HA carrier | 1 |
| Type 1C | 63 |
| Total | 153 |
| Type 2 | |
| Type 2A | 73 |
| Type 2B | 52 |
| Type 2M | 24 |
| Type 2M-C | 2 |
| Type 2N | 9 |
| Type 2N carrier | 1 |
| Type 2N/type 1 | 4 |
| Total | 165 |
| Type 3 | |
| Total | 40 |
| Overall total | 737 |
Sample distribution by VWD type.
HA, hemophilia A.
VWF sequencing and ab1 file processing
VWF Sanger sequencing of all exons, including intron-exon boundaries and ∼50 to 100 bp of intronic sequence, was performed on all 737 unrelated probands at either the Harvard Partners Genome Center or Versiti Blood Center of Wisconsin using the VWF reference sequence NM_000552.22 Ab1 files were processed using Mutation Surveyor (https://softgenetics.com/mutationSurveyor.php), which aligns and calls sequence variants, and all variants were visually confirmed.
Rare variant analysis
To obtain gnomAD frequencies and combined annotation-dependent depletion (CADD) scores for all variants, annotation was performed using SeattleSeq (https://snp.gs.washington.edu/SeattleSeqAnnotation153/index.jsp).23 To be included in these analyses, variants must be nonsynonymous and rare (≤1%) in all gnomAD populations. In the present sample, 650 nonsynonymous rare variants were left after filtering. Box plots were created using the ggplot2 package in R. Linear regressions were also run in R. All subtypes of VWD types 1 and 2 were combined into simply type 1 and type 2 to increase power.
Results
Relationship between VWF:Ag levels, number of rare nonsynonymous variants, ISTH BAT score, and known pathogenic variants
When plotting the relationship between the number of rare nonsynonymous variants per individual and VWF:Ag levels, we observed a statistically significant inverse relationship (P = 1.62 × 10−21). After controlling for age, sex, blood type, and self-reported race, 16.4% of the variance in VWF:Ag levels was explained by the number of rare nonsynonymous variants. Approximately an equal amount of variance in VWF:Ag levels, 16.2%, is explained by ISTH BAT score (P = 1.1 × 10−26). When both the number of rare nonsynonymous variants in VWF per individual and ISTH BAT score are included in the model, 23.8% of the variance in VWF:Ag levels can be explained. The more rare nonsynonymous variants, the lower an individual’s VWF:Ag levels were on average (Figure 1; Table 2). Interestingly, the variance in VWF:Ag levels was observed across but not within VWD types, showing that, in our sample, VWD type is correlated with VWF:Ag levels. This is necessarily the case for the patients with low VWF and type 1 VWD because they were diagnosed in part based on their VWF:Ag levels. However, patients with low VWF and type 1 VWD are defined by additional assays including VWF:RCo and therefore not all have expected VWF:Ag levels for type.

Rare nonsynonymous VWF variants and VWF:Ag levels. The number of rare nonsynonymous VWF variants is a significant predictor of VWF:Ag levels, independent of VWD type (P = 1.62 × 10−21).
Rare nonsynonymous VWF variants and VWF:Ag levels. The number of rare nonsynonymous VWF variants is a significant predictor of VWF:Ag levels, independent of VWD type (P = 1.62 × 10−21).
Variable stratification by number of rare nonsynonymous variants
| Rare nonsynonymous variant count | N | Geometric mean VWF:Ag | Median ISTH BAT score | % with CADD >20 |
|---|---|---|---|---|
| 0 | 322 | 65.6 | 2 | 2.8 |
| 1 | 263 | 37.9 | 5 | 11.8 |
| 2 | 106 | 27.2 | 7.5 | 17.9 |
| 3 | 32 | 35.7 | 6.5 | 12.5 |
| 4 | 14 | 25.7 | 11 | 50.0 |
| >4 | 3 | 25.4 | 1 | 67 |
| Rare nonsynonymous variant count | N | Geometric mean VWF:Ag | Median ISTH BAT score | % with CADD >20 |
|---|---|---|---|---|
| 0 | 322 | 65.6 | 2 | 2.8 |
| 1 | 263 | 37.9 | 5 | 11.8 |
| 2 | 106 | 27.2 | 7.5 | 17.9 |
| 3 | 32 | 35.7 | 6.5 | 12.5 |
| 4 | 14 | 25.7 | 11 | 50.0 |
| >4 | 3 | 25.4 | 1 | 67 |
Geometric mean of VWF:Ag, and median ISTH BAT score and the percentage with CADD >20 stratified by each rare nonsynonymous variant count.
We next modeled the effect of nonpathogenic variants, that is, rare nonsynonymous variants that were either novel, predicted benign, or variants of unknown significance (VUS) by including the number of pathogenic variants as a covariate in a linear regression predicting VWF:Ag level. For the present analyses, pathogenic variants were defined as variants listed in ClinVar as pathogenic or likely pathogenic, or loss-of-function variants due to stop-gain or frameshift. We find that both the number of pathogenic VWF variants harbored by an individual (P = 6.6 × 10−19) and the number of nonpathogenic variants (P = 1.1 × 10−16) significantly predicted VWF:Ag levels. Additionally, we performed a stratified analysis and observed a significant correlation between the number of nonpathogenic variants and VWF:Ag levels among patients with 0 pathogenic VWF variants (N = 603) (P = 2.4 × 10−16). We find that 7.3% of the variance in VWF:Ag levels is explained by pathogenic VWF variants, and 6.1% of the variance in VWF:Ag levels is explained by nonpathogenic variants. By conditioning on all significant predictive factors other than VWF rare variants (sex, blood type, and self-reported race), we find that the number of rare nonsynonymous variants in VWF explains an additional 16.4% of the variance in VWF:Ag (P = 1.3 × 10−51). Together, our model is able to explain 31.1% of the variance in VWF:Ag levels.
Number of rare nonsynonymous variants correlates with VWD type
We then calculated the average number of rare nonsynonymous variants per person, stratified by VWD type. This graph has a correlation coefficient (R2) of 0.89 (Figure 2) (2.4 × 10−13 < P < 3.5 × 10−60 for all VWD types). Healthy controls had only 0.16 rare variants per person on average, whereas that figure quadrupled in patients with low VWF to 0.67 rare nonsynonymous variants per person, and then doubled in VWD type 1 to 1.26 rare nonsynonymous variants per person. VWD types 2 and 3 had the most, with ∼1.5 rare nonsynonymous variants per person on average for each type.
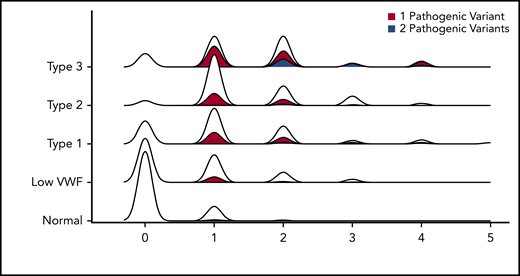
Number of rare nonsynonymous VWF variants per person stratified by VWD type. Density plot of the relationship between number of rare nonsynonymous variants per person and VWD type. Each density plot was colored according to the proportion composed of individuals with either 1 (red) or 2 pathogenic (blue) VWF variants. No person with VWD had >2 pathogenic variants.
Number of rare nonsynonymous VWF variants per person stratified by VWD type. Density plot of the relationship between number of rare nonsynonymous variants per person and VWD type. Each density plot was colored according to the proportion composed of individuals with either 1 (red) or 2 pathogenic (blue) VWF variants. No person with VWD had >2 pathogenic variants.
We also performed association testing between the number of rare nonsynonymous variants in individuals with normal VWF levels and low VWF as well as all types of VWD. All VWD types had significant numbers of rare nonsynonymous variants compared with healthy individuals (Table 3). Whereas 88% of healthy controls had no rare VWF variants, only 52% of patients with low VWF and 26% of patients with VWD type 1 had no rare nonsynonymous variant in the VWF gene, whereas only 5% and 15% of patients with VWD type 2 and 3 had no rare nonsynonymous sequence variant in VWF, respectively (supplemental Figure 1). The observed association between the number of VWF nonsynonymous variants and VWF antigen level was not due solely to the presence of patients with type 3 VWD in the data set who are expected to harbor 2 pathogenic variants because the association remained when we stratified to only include patients with low VWF and type 1 VWD (P = 2.36 × 10−6). Other genetic explanations of pathological bleeding must therefore be sought in these patients. For example, here, we are focusing on rare nonsynonymous variants, however, 3 of the patients with type 3 VWD that had no rare nonsynonymous variants are known to have intronic/splice-site mutations that would not have been included here. Additionally, others could have large-scale deletions or duplications that affect protein function that are not detected with Sanger sequencing.
Variable stratification by VWD type
| VWD type | N | Geometric mean VWF:Ag | Median ISTH BAT score | % with CADD >20 | Average rare nonsynonymous variants per person | P vs healthy |
|---|---|---|---|---|---|---|
| Healthy controls | 210 | 104.8 | 1 | 0 | 0.16 | — |
| Low VWF | 169 | 45.7 | 4 | 13 | 0.67 | 2.4 × 10−13 |
| Type 1 | 153 | 17.9 | 5 | 18 | 1.26 | 1.6 × 10−27 |
| Type 2 | 165 | 45.1 | 9 | 40 | 1.47 | 3.5 × 10−60 |
| Type 3 | 39 | 6.2 | 15 | 55 | 1.5 | 2.7 × 10−34 |
| VWD type | N | Geometric mean VWF:Ag | Median ISTH BAT score | % with CADD >20 | Average rare nonsynonymous variants per person | P vs healthy |
|---|---|---|---|---|---|---|
| Healthy controls | 210 | 104.8 | 1 | 0 | 0.16 | — |
| Low VWF | 169 | 45.7 | 4 | 13 | 0.67 | 2.4 × 10−13 |
| Type 1 | 153 | 17.9 | 5 | 18 | 1.26 | 1.6 × 10−27 |
| Type 2 | 165 | 45.1 | 9 | 40 | 1.47 | 3.5 × 10−60 |
| Type 3 | 39 | 6.2 | 15 | 55 | 1.5 | 2.7 × 10−34 |
Geometric mean of VWF:Ag and median ISTH BAT score, the percentage with CADD >20, and average number of rare nonsynonymous variants per person, stratified by VWD type. P values are comparing the average number of rare nonsynonymous variants in each type of VWD to healthy individuals.
—, no comparison.
Predicted deleterious VWF variants do not fully explain the association between VWF variant burden and VWF:Ag levels
The impact of rare nonsynonymous variants beyond known pathogenic variants in VWF was determined by examining predicted deleterious variants. The CADD score integrates multiple annotations into 1 metric of deleteriousness (eg, a reduction in fitness), contrasts fixed alleles with simulated variants to obtain a score,24 and is an effective way of prioritizing causal variants in genetic analyses. A CADD score of >20 is widely used as a cutoff for predicting pathogenic variants.25 This cutoff represents the level at which only 1% of variants have scores >20. Therefore, we first determined the proportion of patients with each VWD type that had at least 1 variant with CADD >20. In healthy controls, we observed no variants with a CADD >20. However, patients with low VWF and VWD type 1 both had similar proportions of individuals with a variant with CADD >20 (13% to 18%). VWD types 2 and 3 had much higher proportions, at 40% and 55%, respectively (Table 3).
We then compared VWF:Ag levels in 2 groups: those with a variant with CADD >20 and those without. There is a significant difference in VWF:Ag levels between the 2 groups (P = 9.2 × 10−4), with those who have a variant with a CADD >20 having significantly lower VWF:Ag levels than those who do not. When we analyzed differences in VWF:Ag levels between healthy individuals, those with a known pathogenic variant, those with no VWF variant, and those with and without variants with a CADD score >20, it was clear that those with known and predicted pathogenic variants had lower VWF:Ag levels than healthy individuals, those with no VWF variant, and those with a variant with a CADD score <20 (Figure 3).
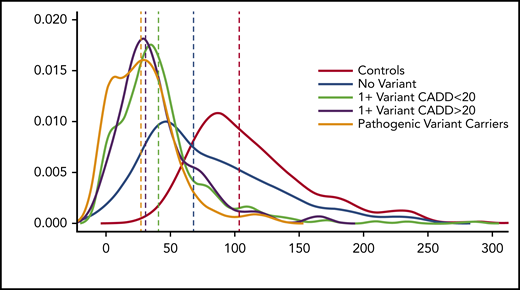
VWF:Ag levels stratified by predicted or known pathogenic VWF variants. Density plot of VWF:Ag levels stratified by carrier status of different variant types.
VWF:Ag levels stratified by predicted or known pathogenic VWF variants. Density plot of VWF:Ag levels stratified by carrier status of different variant types.
Variants with CADD scores >20 explained an additional 1% in variation in VWF:Ag levels on top of known pathogenic variants, but additional variation could still be explained by rare variants with CADD scores <20, making prediction of which variants will affect VWF:Ag levels difficult. When we included both variants with CADD score >20 and all nonpathogenic variants (ie, VUS) in a linear regression model, the number of rare VUS harbored by an individual was a better predictor than whether they carried a variant with CADD >20. Overall, our results suggest that rare VWF coding variants currently deemed VUS, many with CADD scores <20, contribute significantly to VWD and VWD disease severity as measured by VWF:Ag levels and ISTH BAT score.
Discussion
A clinical diagnosis of VWD is made by a combination of low VWF:Ag levels and bleeding. The genetic confirmation of this diagnosis is more definitive in cases of VWD type 2, VWD type 3, and severe VWD type 1 as rare genetic variants have been associated with these types and many of the variants have been adequately curated through functional studies in cell and animal models as well as being replicated in family and cohort studies.26 However, milder forms of VWD such as VWD type 1 and low VWF have been difficult to characterize genetically as VWF is a large, highly polymorphic gene and only 40% of these patients showed a rare damaging variant in previous studies.6
In our study, we have shown that we can explain a significant percentage of variance in VWF:Ag levels using rare nonsynonymous variants. The number of rare nonsynonymous variants present in the VWF gene is significantly associated with the VWF:Ag levels of the patients, and this association was observed across, rather than within, VWD types. This association between VWF:Ag levels and number of rare nonsynonymous variants remains highly significant even when controlling for known pathogenic VWF variants, suggesting that other variants exist in VWF and elsewhere in the genome that contribute to VWF:Ag levels. We assume that a proportion of the VUS are pathogenic, but we do not have enough evidence to definitively state that at this time. Furthermore, each VWD type (including low VWF) was significantly associated with number of rare variants when compared with healthy individuals. Lastly, we showed that a higher proportion of predicted pathogenic variants is present in the VWF gene of patients with VWD types 2 and 3. It is difficult to say whether the number of rare nonsynonymous variants is having an additive effect on the reduction of VWF:Ag levels or that having more variants simply increases the chances that 1 of those variants affects VWF:Ag level, as current data are insufficient to distinguish between these 2 possibilities.
It is perhaps expected that VWD types 2 and 3 will have on average a greater proportion of known and predicted pathogenic variants, as defined CADD >20, as these types of VWD are known to be caused mainly by mutations in the VWF gene, and VWD type 3 necessarily requires 2 distinct disruptions to VWF. However, when taking the VWD type agnostic approach and simply comparing VWF:Ag levels in those with and without a variant with a CADD >20, our results show that VWF:Ag levels on their own predict the presence of a predicted pathogenic variant in the VWF gene, with those patients with lower VWF:Ag levels having a significantly greater chance of harboring a predicted pathogenic VWF variant. Furthermore, when in the same linear regression, the number of rare nonsynonymous variants and the presence of a variant with CADD >20 were both significant predictors of VWF:Ag levels (P = 1.5 × 10−4 and .02, respectively).
Taken together, this strongly suggests that differences in VWF:Ag levels are due in large part to both number of rare nonsynonymous variants in the VWF gene and presence of a known or predicted pathogenic VWF variant. This is somewhat expected as the chances of having a pathogenic variant, either known or yet uncharacterized, increase in direct relation to the number of rare nonsynonymous variants present. However, even among patients with known pathogenic mutations, their VWF:Ag levels could not be fully explained by the presence of these mutations. Therefore, in the future, all patients with VWD could benefit from whole-exome or whole-genome sequencing in order to understand additional factors affecting their disease susceptibility.
Our analyses also provide further proof that VWD type 1 and low VWF are not monogenic traits, with many other genetic factors likely contributing to the variation in VWF:Ag levels. In fact, there are 5 previously published genome-wide association studies of VWF:Ag levels13-17 that together have identified variants in over 2 dozen genes besides VWF and ABO that are associated with the phenotype. Several of the variants were functionally validated in vitro using gene silencing in cultured endothelial cells.14 In a review of the genetics of VWF:Ag levels, Swystun and Lillicrap conclude that future studies looking specifically at the influence of rare variants on VWF:Ag levels through whole-genome/exome analysis are needed to increase understanding of the genetic basis of pathological quantitative abnormalities of VWF:Ag levels. These types of analyses would include elucidation of effects of expressivity, penetrance, epistasis, and gene-environment influences.10 They further postulate that these types of analyses will improve the molecular diagnosis of VWD type 1 and low VWF.
Unlike common variant associations in which large sets of highly correlated single-nucleotide polymorphisms spanning many kilobases of the genome are often implicated in disease risk, rare variant association analysis has the power to implicate specific variants and genes, enabling more rapid translation of findings into treatments. These results of rare nonsynonymous variant analysis show that it is likely that patients with VWD that have higher VWF:Ag levels are the least likely to get a definitive answer for the cause of their VWD from sequencing their VWF gene alone.
There were 7 patients with VWD type 3 in this study with absent VWF:Ag levels who did not have a rare nonsynonymous variant in VWF. As mentioned previously, Sanger sequencing data will not detect all large copy-number variations, or intronic variations, and thus it is possible that these individuals have VWF variants we were unable to detect. This is not an issue with next-generation sequencing. Another potential limitation of our study is the lack of sequencing of the parents of these cases, as we cannot determine whether multiple variants occur on the same allele. In the future, sequencing of parents would allow us to determine whether multiple variants are occurring on the same or different chromosomes. This would enable us to more accurately predict VWD and antigen level from genetic information alone, as 2 variants on the same allele could only have a partial reduction in antigen level compared with having pathogenic variants on both alleles, which could lead to VWD type 3 and near absent antigen levels.
An interesting finding from our data set is that the ISTH BAT predicted VWF:Ag levels approximately as well as the number of rare nonsynonymous variants, indicating that patients that present to clinic with low VWF levels and bleeding are the ones most likely to have rare variants in VWF. This has implications for VWD classification, as patients with low VWF and type 1 VWD have very few rare nonsynonymous variants in VWF but higher than healthy controls, therefore indicating that their milder phenotype may be explained by modifier variants elsewhere in the genome. VWF:Ag level and ISTH BAT are both confounded with VWD type and with each other as VWD type is defined by these 2 variables. Our ultimate goal is to better predict who is at risk for bleeding from genetic information before they have potentially life-threatening complications. Genetic factors are not dependent on phenotype (VWF:Ag level, ISTH BAT score, VWD type, etc) and therefore are necessarily the upstream effector of all of these correlated traits. Additionally, it is well known that a significant amount of variance in VWF:Ag level can be predicted using measured environmental factors (age, sex, blood type, and self-reported race) and that including these in the model allowed for more accurate VWF:Ag level prediction when combined with genetic information. Ideally, through genetic diagnosis, we can relatively accurately predict each of these traits before their clinical consequences are known and take the proper precautions to avoid unnecessary harm to patients and to provide care as early as possible.
In summary, our study shows that rare variants in VWF account for a significant component of the VWF-level variability in patients diagnosed with VWD. Our study paves the way for a more genetically oriented classification system for patients with VWD.
For original data, please contact Pamela A. Christopherson at pchristopherson@versiti.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgment
This work was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute for the Zimmerman Program and multiple investigators (HL081588, HL112614, and HL144457).
Authorship
Contribution: B.S. and G.H. conceived of the manuscript, performed all sequence and statistical analyses, and made all figures; B.S., P.A.C., G.H., and J.D.P. wrote the manuscript; P.A.C. collected Zimmerman Program subject information; R.R.M. is the leader of the Program Project Grant and supervised the Zimmerman Program subject classification; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: R.R.M. is a consultant for AstraZeneca, Baxter, Bayer, Biogen Idec, CSL Behring, Grifols, Immucor, and Octapharma. The remaining authors declare no competing financial interests.
A complete list of Zimmerman Program Investigators appears in the supplemental Appendix.
Correspondence: Jorge Di Paola, Department of Pediatrics, Washington University in St Louis, 660 S Euclid Ave, Campus Box 8208, St Louis, MO 63110; e-mail: dipaolaj@wustl.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal