

In this issue of Blood, 1 report abnormal retention of mitochondria in circulating erythrocytes and the presence of mitochondrial DNA (mtDNA) in plasma in sickle cell disease (SCD). Furthermore, they identify cell-free mtDNA (cf-mtDNA) as a danger-associated molecular pattern (DAMP) in SCD. Although mtDNA has been known as a DAMP molecule for some time,2 this interesting study showed that SCD increases cell-free DNA (cfDNA) and cf-mtDNA at baseline and crisis in humans and Townes sickle mice (see figure).
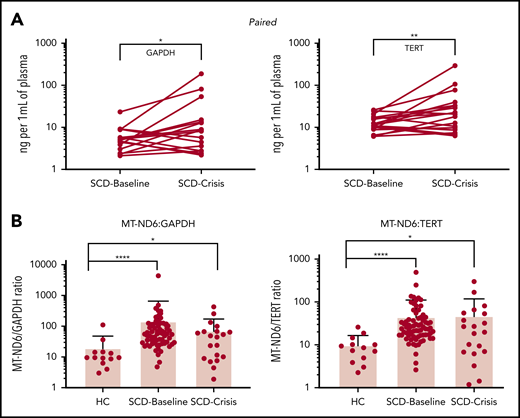
(A) Paired samples: SCD-Baseline and SCD-Crisis pairs (n = 18). Circles represent the mean of each sample. *P < .05, **P < .01, nonparametric Wilcoxon matched-pairs signed-rank test. (B) Quantitation of cf-mtDNA/cf-nDNA ratio using the following mitochondrial and nuclear targets: MT-ND1/GAPDH, MTND1/TERT, MT-ND6/GAPDH, and MT-ND6/TERT. Error bars represent the sample mean ± standard deviation. *P < .05, ***P < .0005, ****P < .0001, nonparametric Kruskal-Wallis test with Dunn’s multiple-comparison test. cf-nDNA, cell-free nuclear DNA. See the complete Figure 1 in the article by Tumburu et al that begins on page 3116.
(A) Paired samples: SCD-Baseline and SCD-Crisis pairs (n = 18). Circles represent the mean of each sample. *P < .05, **P < .01, nonparametric Wilcoxon matched-pairs signed-rank test. (B) Quantitation of cf-mtDNA/cf-nDNA ratio using the following mitochondrial and nuclear targets: MT-ND1/GAPDH, MTND1/TERT, MT-ND6/GAPDH, and MT-ND6/TERT. Error bars represent the sample mean ± standard deviation. *P < .05, ***P < .0005, ****P < .0001, nonparametric Kruskal-Wallis test with Dunn’s multiple-comparison test. cf-nDNA, cell-free nuclear DNA. See the complete Figure 1 in the article by Tumburu et al that begins on page 3116.
The evidentiary support for their core observations is considerable. Healthy mature red blood cells do not contain mitochondria, except in rare disease conditions, such as Rett syndrome.3 The investigators used multiple techniques to demonstrate that SCD increases mitochondrial retention in the sickle erythrocyte. These data provide intriguing support for the idea that the sickle erythrocyte is likely the primary cellular source for circulating cf-mtDNA.
The investigators quantified and sequenced cf-DNA, as well as determined the effects of DNA subtypes on neutrophil extracellular trap (NET) formation, to explore the role of cf-mtDNA in SCD. They demonstrated that SCD increases cf-mtDNA levels approximately five fold to 10-fold at baseline compared with levels in healthy controls. However, the mechanism(s) by which cf-mtDNA causes and quantitatively contributes to inflammation in SCD patients remains unclear. Although the investigators focused attention on NET formation, additional studies will be required to clarify this issue. For example, we and other investigators have compared the effects of SCD on relative changes in another DAMP: high mobility group box-1 (HMGB1). It was reported that SCD increases HMGB1 by approximately threefold4 and ∼1.8-fold5 in patients at baseline relative to healthy controls. These data are consistent with the results presented by Tumburu et al. Interestingly, however, cf-mtDNA in SCD patients in crisis does not change relative to cf-mtDNA in SCD patients at baseline. In contrast, HMGB1 in SCD patients in crisis increases by approximately twofold4 and ∼1.7-fold5 compared with their levels at baseline. If inflammation is proportional to the level of DAMPs released, then the observation that cf-mtDNA did not increase during crisis (see figure) requires further exploration. For example, could cf-mtDNA have increased locally in tissue beds most severely damaged during crisis?
Alternatively, DAMPs come in all shapes and sizes, and the effects of physical and biochemical differences can be exquisitely nuanced. Accordingly, if the cf-mtDNA released during crisis is different from the cf-mtDNA released at baseline, such a biochemical change in the DAMP may alter biological activity. Because DNA methylation plays an important role in epigenetics, the investigators analyzed cf-mtDNA for any detectable differences. They showed that cf-mtDNA released during crisis was more hypomethylated than was cf-mtDNA released by SCD patients at baseline and even more hypomethylated than cf-mtDNA released by healthy controls. This is an intriguing observation that the investigators build on when they compared the effects of genomic DNA or platelet mtDNA in RPMI 1640 media with healthy human plasma and SCD plasma, which contained high concentrations of cf-mtDNA on NET formation. Their data clearly showed that SCD plasma increased NETosis. However, because experimental conditions were not directly comparable, they cautiously speculated that cf-mtDNA in plasma from SCD patients increased NET formation, possibly because it was hypomethylated. Future studies using comparable conditions may help to determine the relative contributions of this growing list of DAMPs on inflammation and NETosis in SCD at baseline and crisis. Indeed, findings from such studies may explain why crisis worsens, even though cf-mtDNA did not increase beyond baseline.
Findings from this report will likely spur the further studies needed to unravel the complex processes by which sickle erythrocytes increase inflammation beyond the damage caused to the vessel wall by sickled erythrocytes. If cf-mtDNA can be specifically targeted and inflammation is reduced in SCD mice, then cf-mtDNA may be an important mediator of inflammation that increases vaso-occlusion by what the investigators suggest is neutrophil activation and NETosis. Direct comparisons of DAMPs in SCD will reveal whether cf-mtDNA is an important mediator of inflammation and vasocongestion or just another biomarker of inflammation.
Conflict-of-interest disclosure: K.A.P. is a founder and member of ReNeuroGen LLC, an early-stage virtual pharmaceutical company developing therapies for the treatment of neuroinflammation in multiple sclerosis, bronchopulmonary dysplasia, vasculopathy in sickle cell disease, ischemic stroke, silent cerebral infarct, and traumatic brain injury. K.R.R. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal