

Key Points
Heightened activation of eMks/platelets cause formation of aneurysms in developing brains of mice lacking PDPN.
PDPN lessens collagen-1–induced secretion of angiopoietin-1 from eMks, preventing detrimental TIE-2 activation in sprouting endothelium.

Abstract
During early embryonic development in mammals, including humans and mice, megakaryocytes (Mks) first originate from primitive hematopoiesis in the yolk sac. These embryonic Mks (eMks) circulate in the vasculature with unclear function. Herein, we report that podoplanin (PDPN), the ligand of C-type lectin-like receptor (CLEC-2) on Mks/platelets, is temporarily expressed in neural tissue during midgestation in mice. Loss of PDPN or CLEC-2 resulted in aneurysms and spontaneous hemorrhage, specifically in the lower diencephalon during midgestation. Surprisingly, more eMks/platelets had enhanced granule release and localized to the lower diencephalon in mutant mouse embryos than in wild-type littermates before hemorrhage. We found that PDPN counteracted the collagen-1–induced secretion of angiopoietin-1 from fetal Mks, which coincided with enhanced TIE-2 activation in aneurysm-like sprouts of PDPN-deficient embryos. Blocking platelet activation prevented the PDPN-deficient embryo from developing vascular defects. Our data reveal a new role for PDPN in regulating eMk function during midgestation.
Introduction
During early development in mammals, a wave of primitive hematopoiesis occurs within blood islands of the yolk sac, before the emergence of definitive hematopoiesis that relies on self-renewed multipotent hematopoietic stem cells (HSCs).1 In mice, primitive hematopoiesis begins at approximately embryonic day 7.5 (E7.5) and generates only erythroid progenitors, embryonic macrophages, and Mks.2-4 Erythroid progenitors and embryonic macrophages produce nucleated fetal erythrocytes, to transport oxygen and nutrients, and tissue macrophages to support the fast-growing embryo.5 Embryonic Mks (eMks) are found in the yolk sac and throughout the vasculature of embryos during midgestation (E10-E13).6,7 These eMks are primarily diploid and produce platelets in the vasculature. However, the function of these eMks/platelets remains unclear.
Coinciding with the appearance of circulating eMks, angiogenesis begins rapidly extending into the brain from the perineural vascular plexus (PNVP). Angiogenic sprouting into the brain is primarily driven by neural cell–derived vascular endothelial growth factor (VEGF),8 as well as other growth factors, including angiopoietin-1 (ANGPT-1).9,10 Deficiencies of these factors results in defective vascular development,11,12 although increased expression of the ANGPT-1 endothelial receptor TIE-2 or enhanced response to ANGPT-1, leads to hemangioma and venous malformations.13,14 Adult platelets contain large quantities of these in their granules,15,16 but whether and how eMks regulate angiogenesis in the brain during midgestation is unclear.
The neuroepithelium expresses the O-glycoprotein podoplanin (PDPN) highly during mouse embryonic development.17,18 The loss of PDPN (Pdpn−/−) in mice leads to spontaneous hemorrhaging in midgestation brains.19 PDPN is the only physiological ligand of C-type lectin-like receptor 2 (CLEC-2) on Mks/platelets. Mice lacking CLEC-2 (Clec-2−/−) also develop brain bleeding that resembles bleeding in Pdpn−/− embryos.20 However, the nature of vascular defects and how Mks/platelets contribute to it in the absence of PDPN-CLEC-2 remains unclear.
In the present study, we found that eMks express CLEC-2 and that loss of PDPN or CLEC-2 results in reduced angiogenic sprouting and aneurysms within the diencephalon during midgestation in mice. Unexpectedly, loss of PDPN or CLEC-2 caused an increased localization of eMks/platelets within the PNVP and aneurysm-like angiogenic sprouts in the diencephalon. We found that these eMks/platelets were active and released ANGPT-1 to induce TIE-2 activation in angiogenic sprouts, leading to formation of aneurysms. These results reveal a new role for PDPN in the control of eMk activation during midgestation.
Methods
This section contains a brief description of the main methods. An expanded description of the methods is available in the supplemental Data, available on the Blood Web site.
Mice
All mice were maintained, and experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of Oklahoma Medical Research Foundation. Timed-mated females were obtained from natural matings by crossing males with females of breeding age. The presence of a copulatory plug denoted E0.5.
Histology, immunostaining, and image acquisition
Mouse embryos were collected and photographed at dissection. For histology, embryos were fixed in 10% formalin, and 5-μm paraffin-embedded coronal sections were cut and stained with hematoxylin and eosin. For immunofluorescence, tissues were fixed in 4% paraformaldehyde and cryosections were cut at a thickness of 20 to 100 μm, blocked, incubated with primary and then secondary antibodies, counterstained with nuclear marker, and mounted for imaging with a Zeiss LSM710 microscopy system.
Transmission electron microscopy
Samples of embryonic heads were prepared as previously described.21 Blocks were cut at 1 μm for semithin sections and 100 nm for ultrathin grids. Toluidine blue O–stained semithin sections were imaged with a Nikon Eclipse E600 microscope with a 100× oil immersion objective (Apochromat; numeric aperture, 1.40). Ultrathin grids were stained with uranyl acetate and imaged with a Hitachi H-7600 transmission electron microscope equipped with a Kodak digital camera. Ultrastructural images were layered with pseudocolors using Canvas Draw.
PDPN- and/or collagen-1–induced eMk secretion assay
Fetal Mks were cultured as previously reported.22 Plates were coated with recombinant mouse PDPN/human IgG Fc chimera (PDPN, 5 μg/mL), type 1 collagen (COL-1, 20 μg/mL), or both. Cells were plated at 37°C for 30 minutes and adherent Mks were fixed. For inhibitory experiments, eMks were treated with 30 μmol ticagrelor and 10 μmol aspirin for 30 minutes at room temperature before plating.
Data analysis and statistics
Quantification and statistical analyses are reported where applicable in the figure legends and supplemental Data. Statistical analyses were performed in GraphPad Prism 7. An analysis of variance (ANOVA) with Dunnett’s post hoc test was used when comparing experimental groups with the control, and Tukey’s test was used when comparing experimental groups against each other.
Results
Mice lacking PDPN or CLEC-2 develop aneurysms in the diencephalon during midgestation
PDPN is expressed in the brain during embryonic development23 ; however, its expression pattern was unclear. Our immunostaining results showed that PDPN was temporally expressed on neuroepithelium during midgestation (Figure 1A; supplemental Figure 1A). Histology showed that brains of embryos lacking PDPN or CLEC-2 (Pdpn−/− or Clec-2−/−) exhibited aneurysms growing into the third ventricle during this developmental stage, relative to the wild-type (WT). Some aneurysms ruptured in the parenchyma and into the ventricles (Figure 1A-D; supplemental Figure 1B), with no change in lower diencephalon perfusion (supplemental Figure 1C). Spontaneous hemorrhaging primarily occurred in the diencephalon, with full penetrance by E12.5 (Figure 1A-B; supplemental Figure 1D). Early in the bleeding phenotype, these aneurysm-like sprouts in the diencephalon were tenuous and full of red blood cells (Figure 1D; supplemental Videos 1-2), suggesting that ruptured aneurysms were the primary source of hemorrhaging.
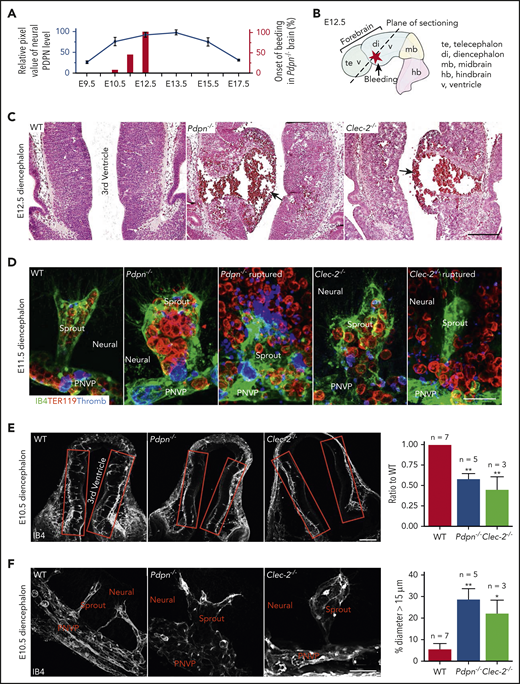
Mice lacking PDPN or CLEC-2 develop aneurysms and spontaneous hemorrhages in the diencephalon at midgestation. (A) Quantification of PDPN expression from confocal images of mouse embryonic diencephalon, with values representing 3 experiments. Bars represent percentages of bleeding onset in the Pdpn−/− diencephalon. (B) The E12.5 mouse brain. (C) Representative images of hematoxylin and eosin–stained embryonic coronal sections of the diencephalon. Arrows indicate aneurysms. (D) Confocal images of lower diencephalon aneurysm-like radial sprouts. IB4 marks the vasculature. TER119 marks erythrocytes. Thromb marks eMks and platelets. Neural, brain tissue. (E) Confocal images of angiogenic sprouting into the diencephalon (red boxes). The bar graph on the right represents the ratio of radial sprouts in the mutant diencephalon compared with the paired WT. (F) Confocal images of aneurysm-like sprouts in the diencephalon. Bar graph on the right represents the percentage of dilated sprouts (>15 μm in diameter) throughout mutant diencephalons relative to WT. Scale bars, 100 μm (C,E); 25 μm (D); and 20 μm (F). Data are means ± standard error of the mean; n, number of embryos. *P < .05; **P < .01.
Mice lacking PDPN or CLEC-2 develop aneurysms and spontaneous hemorrhages in the diencephalon at midgestation. (A) Quantification of PDPN expression from confocal images of mouse embryonic diencephalon, with values representing 3 experiments. Bars represent percentages of bleeding onset in the Pdpn−/− diencephalon. (B) The E12.5 mouse brain. (C) Representative images of hematoxylin and eosin–stained embryonic coronal sections of the diencephalon. Arrows indicate aneurysms. (D) Confocal images of lower diencephalon aneurysm-like radial sprouts. IB4 marks the vasculature. TER119 marks erythrocytes. Thromb marks eMks and platelets. Neural, brain tissue. (E) Confocal images of angiogenic sprouting into the diencephalon (red boxes). The bar graph on the right represents the ratio of radial sprouts in the mutant diencephalon compared with the paired WT. (F) Confocal images of aneurysm-like sprouts in the diencephalon. Bar graph on the right represents the percentage of dilated sprouts (>15 μm in diameter) throughout mutant diencephalons relative to WT. Scale bars, 100 μm (C,E); 25 μm (D); and 20 μm (F). Data are means ± standard error of the mean; n, number of embryos. *P < .05; **P < .01.
To further determine whether defective angiogenesis occurs before hemorrhage, we analyzed angiogenic sprouting at E10.5. Our results showed a significant reduction in nascent radial sprouts into the diencephalon from the PNVP (Figure 1E), many of which were aneurysm-like (Figure 1F) in both Pdpn−/− and Clec-2−/− embryos. WT angiogenetic sprouts (supplemental Figure 1E) and Pdpn−/− aneurysm-like sprouts had either no (E10.5) or similar (E11.5) coverage of pericytes (supplemental Figure 1F-H), which indicates that defective angiogenesis is not initially caused by impaired vascular maturation. Together, these results revealed that PDPN and CLEC-2 are critical for angiogenic sprouting in the diencephalon at midgestation in mice.
Increased localization of eMks in aneurysm-like sprouts and PNVP of the Pdpn−/− and Clec-2−/− lower diencephalons
Platelet CLEC-2 interaction with PDPN is critical for vascular integrity in adult mice.24 To investigate the role of platelets in angiogenic sprouting at midgestation, we stained the Pdpn−/− and Clec-2−/− diencephalons with an antibody to CD41 that is specifically expressed on Mks/platelets. Unexpectedly, we found an increased number of large CD41+ cells localized in the abnormal sprouts and PNVP of the lower diencephalons of both Pdpn−/− and Clec-2−/− brains at E11.5 (Figure 2A-B). As indicated by an Mk-specific nuclear enhanced green fluorescent protein reporter, which was generated by breeding ROSA nT-nG mice,25 with the Mk-specific Cre recombinase mouse line driven by Pf4 promoter,26 these nucleated CD41+ cells were Mks (Figure 2C). Flow cytometry and imaging analyses indicated that these circulating CD41+ cells were diploid and expressed ITGA2B (CD41), GP1BB (CD42c), ITGA2 (CD49b), GPVI (GP6), and CLEC-2, which are common molecules on Mks/platelets (Figure 2D-E). In addition, these nucleated CD41+ cells produced proplatelets within the brain vasculature (Figure 2F). To sum up, these data support that these large CD41+ cells were eMks, most likely derived from the first primitive wave of hematopoiesis (Figure 2G).5

Increased localization of eMks within aneurysm-like sprouts and PNVP of Pdpn−/− and Clec-2−/− lower diencephalon. (A) Confocal images of aneurysmlike sprouts with eMks (asterisks) and platelets. CD41 marks eMks and platelets. (B) Confocal images and representative quantified number of eMks and platelets (Plts) in the diencephalon. Yellow boxes indicate the lower diencephalon. CD41 marks eMks and platelets. (C) Micrograph of eMks (asterisks) double labeled with CD41 and nuclear enhanced green fluorescent protein (EGFP) within diencephalon of ntdTomato-R26;Pf4Cre embryonic brain. The tdTomato labeling of other nuclei is pseudocolored blue. (D) Flow cytometric data of E11.5 peripheral blood (PB) stained with anti-CD41 to gate eMks and with Hoechst 33342 for nuclear ploidy. Results represent 6 experiments. (E) Results of flow cytometric analysis of platelets from WT midgestation blood vs adult mouse blood. Receptor levels are represented as relative fluorescence. Data represent the mean ± standard error of the mean; n, number of replicate animals. (F) Confocal image of an eMk with proplatelet formations (arrows) within lower diencephalon vasculature. (G) Diagram showing temporal representation of primitive and definitive hematopoietic waves during embryonic development. Origin of progenitor cells (blue arrows) and location of hematopoiesis (green arrows) are indicated. AGM, aorta-gonad-mesonephros. (H) Graph represents the ratio of eMks to platelets within embryonic PB. Scale bars, 20 μm (A-C); 100 μm (B); and 10 μm (F). **P < .01.
Increased localization of eMks within aneurysm-like sprouts and PNVP of Pdpn−/− and Clec-2−/− lower diencephalon. (A) Confocal images of aneurysmlike sprouts with eMks (asterisks) and platelets. CD41 marks eMks and platelets. (B) Confocal images and representative quantified number of eMks and platelets (Plts) in the diencephalon. Yellow boxes indicate the lower diencephalon. CD41 marks eMks and platelets. (C) Micrograph of eMks (asterisks) double labeled with CD41 and nuclear enhanced green fluorescent protein (EGFP) within diencephalon of ntdTomato-R26;Pf4Cre embryonic brain. The tdTomato labeling of other nuclei is pseudocolored blue. (D) Flow cytometric data of E11.5 peripheral blood (PB) stained with anti-CD41 to gate eMks and with Hoechst 33342 for nuclear ploidy. Results represent 6 experiments. (E) Results of flow cytometric analysis of platelets from WT midgestation blood vs adult mouse blood. Receptor levels are represented as relative fluorescence. Data represent the mean ± standard error of the mean; n, number of replicate animals. (F) Confocal image of an eMk with proplatelet formations (arrows) within lower diencephalon vasculature. (G) Diagram showing temporal representation of primitive and definitive hematopoietic waves during embryonic development. Origin of progenitor cells (blue arrows) and location of hematopoiesis (green arrows) are indicated. AGM, aorta-gonad-mesonephros. (H) Graph represents the ratio of eMks to platelets within embryonic PB. Scale bars, 20 μm (A-C); 100 μm (B); and 10 μm (F). **P < .01.
The ratio of circulating eMks vs platelets in the peripheral blood was much higher during midgestation than at late gestation (Figure 2H). Furthermore, an increased number of these cells were detected specifically in the vasculature of Pdpn−/− and Clec-2−/− lower diencephalons, though not in the midbrains, hindbrains, or limb buds, before hemorrhaging at E10.5, compared with WT littermates (supplemental Figure 2A), suggesting an important role of these eMks in defective angiogenesis.
Primitive hematopoiesis occurs in the yolk sac.1 Interestingly, we found that PDPN was highly expressed in the yolk sac on visceral endodermal cells, which compose a layer of epithelial cells adjacent to the vasculature (supplemental Figure 2B). However, quantification of eMks revealed no significant change in Pdpn−/− or Clec-2−/− yolk sacs relative to that of WT at E10.5. In addition, the absolute number of eMks in the yolk sac and embryo proper was similar between WT and Pdpn−/− (supplemental Figure 2C-D). No obvious morphological abnormalities of vessels or eMks were found in either Pdpn−/− or Clec-2−/− yolk sacs compared with WT (supplemental Figure 2C,E). These results indicate that an increased number of eMks in mutant diencephalons were not a result of increased eMk generation in yolk sacs.
eMks interact with PDPN-expressing neuroepithelium
Pdpn−/− and Clec-2−/− embryos share the same vascular defects and bleeding phenotype, suggesting that interaction of PDPN and CLEC-2 is critical for angiogenesis in the diencephalon during midgestation. Neuroepithelial PDPN levels were high at this stage around the vasculature, especially at the angiogenic front (supplemental Figure 3A). We asked whether neuroepithelial PDPN interacts with circulating eMks. High-resolution confocal imaging indicated that eMks in the PNVP were closely associated with a PDPN+ neuroepithelium in E10.5 embryos (Figure 3A). Transmission electron microscopy (TEM) and confocal microscopy showed that these eMks had lamellipodia, which were devoid of demarcation membrane system and granules (Figure 3B-C), similar to the ectoplasmic ring structure in nonmammalian vertebrate thrombocytes.27,28 Pdpn−/− and Clec-2−/− eMks had lamellipodia similar to those of WT eMks (Figure 3C). The WT eMk lamellipodia processes appeared to interact with the neuroepithelium in the vessels (Figure 3B). However, ultrathin TEM sections are only 0.1 μm thick, which made it technically unfeasible to detect physical interactions between relatively less-abundant eMk lamellipodia and the neuroepithelium. To solve this problem, we used high-powered bright-field microscopy of >10 serial semithin sections (1 μm thickness per section) per eMk, which enabled surveillance of ∼100 times more area than the TEM images (supplemental Video 3). We imaged at least 20 eMks per embryo and found significantly more WT eMks in the PNVP or vascular sprouts that interacted with the neuroepithelium than in Pdpn−/− eMks (Figure 3D), even though there were more eMks present in the Pdpn−/− brain. These results indicate the importance of PDPN in promoting these interactions.

eMks interact with PDPN-expressing neuroepithelium. (A) Confocal microscopic image of single optical slice (left) and a 3-dimensoinal surface rendering (right) of an eMk within the lower diencephalon PNVP closely associated with PDPN+ neural tissue (arrows). (B) Pseudocolored TEM image of an eMk with lamellipodia (yellow arrows) and neuroepithelium (red arrows point to interaction with eMk) in an angiogenic sprout (green) in the lower diencephalon. NEC, neuroepithelial cell. EC, endothelial cell. (C) Confocal microcopy of eMks stained with the α-granule marker TSP-1 in the lower diencephalon. CD41 marks the eMks; arrows mark CD41+TSP-1− ectoplasmic lamellipodia. (D) Representative bright-field microscopy of semithin sections of eMks and their lamellipodia (yellow arrows) in the PNVP (left image) and vascular sprouts in the neural tissue (right 2 images) of the diencephalon. Red arrows mark interactions of lamellipodia processes with neuroepithelium (NEC). The percentage of eMks interacting with NECs in the diencephalon is shown on the right. Data represent mean ± standard error of the mean; n, embryos with at least 20 eMks/embryos analyzed. Scale bars, 10 μm (A,C-D); and 2 μm (B). **P < .01.
eMks interact with PDPN-expressing neuroepithelium. (A) Confocal microscopic image of single optical slice (left) and a 3-dimensoinal surface rendering (right) of an eMk within the lower diencephalon PNVP closely associated with PDPN+ neural tissue (arrows). (B) Pseudocolored TEM image of an eMk with lamellipodia (yellow arrows) and neuroepithelium (red arrows point to interaction with eMk) in an angiogenic sprout (green) in the lower diencephalon. NEC, neuroepithelial cell. EC, endothelial cell. (C) Confocal microcopy of eMks stained with the α-granule marker TSP-1 in the lower diencephalon. CD41 marks the eMks; arrows mark CD41+TSP-1− ectoplasmic lamellipodia. (D) Representative bright-field microscopy of semithin sections of eMks and their lamellipodia (yellow arrows) in the PNVP (left image) and vascular sprouts in the neural tissue (right 2 images) of the diencephalon. Red arrows mark interactions of lamellipodia processes with neuroepithelium (NEC). The percentage of eMks interacting with NECs in the diencephalon is shown on the right. Data represent mean ± standard error of the mean; n, embryos with at least 20 eMks/embryos analyzed. Scale bars, 10 μm (A,C-D); and 2 μm (B). **P < .01.
Heightened activation of eMks and platelets in Pdpn−/− and Clec-2−/− vasculature during midgestation
PDPN activates platelet CLEC-2 through a hemi-immunoreceptor tyrosine–based activation motif (hemITAM) signaling pathway to promote vascular integrity.21,24 We thus hypothesized that loss of PDPN or CLEC-2 would cause reduced activation of eMks and platelets, which would subsequently lead to vascular defects. Unexpectedly, using antibodies specific to either activated Syk or activated PLCγ2,29,30 which are downstream effectors of hem-ITAM signaling, we detected activated Syk and PLCγ2 in the eMks in Pdpn−/− and Clec-2−/− embryos, comparable to that in WT (Figure 4A-B; supplemental Figure 3B-D). We also found comparable integrin αIIbβ3 activation by using the JON/A antibody (supplemental Figure 3E). This result indicates that the defective angiogenesis phenotype is not the result of decreased eMk ITAM signaling.
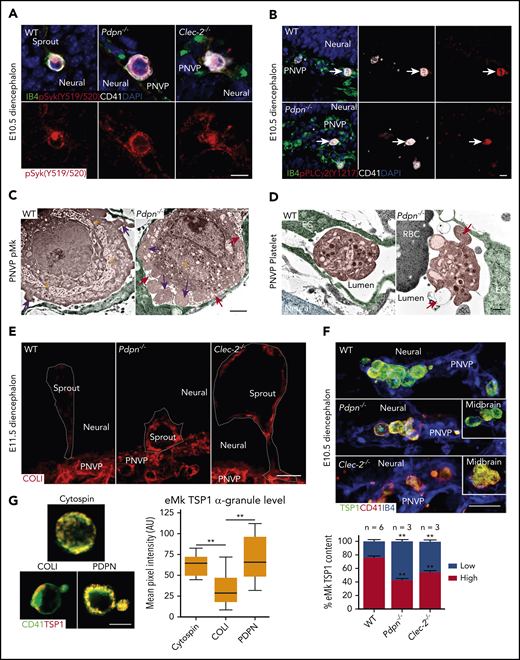
Heightened activation of eMks and platelets in Pdpn−/− and Clec-2−/− vasculature during midgestation. (A-B) Confocal images of diencephalon sections with an antibody to phospho-Syk (Y519/520) (A) or an antibody to phospho-PLCγ2 (Y1217; arrows) (B). 4′,6-Diamidino-2-phenylindole is the nuclear stain. (C-D) Pseudocolored TEM of eMks (C) or platelets (D) in the PNVP vessel lumen of the lower diencephalon. eMk demarcation membrane system (asterisks), microparticles (red arrows), and lamellipodia (pink arrows) are indicated (C). Platelet filopodia projections (orange arrow) and granule secretion (red arrow) are indicated (D). (E) Confocal images of diencephalon sprouts and COL-1 expression. Dashed lines outline each sprout. (F) Confocal images of eMks with α-granules labeled with TSP-1 in the lower diencephalon. Insets show eMks in the midbrain. Bar graph (bottom) represents the percentage of fetal Mks with high or low TSP-1 threshold levels. (G) Confocal images of eMks isolated from E11.5 peripheral blood, that were either plated on a slide through cytospin as a negative activation control or incubated on a slide coated with COL-1 or PDPN for 30 minutes. The α-granules were labeled with TSP-1. The bar graph (right) represents eMk α-granule intensity. Data represent mean ± standard error of the mean and 3 experimental repeats with at least 3 technical repeats each. Scale bars, 10 μm (A-B); 2 μm (C); 500 nm (D); 20 μm (E-F); and 5 μm (G). **P < .01.
Heightened activation of eMks and platelets in Pdpn−/− and Clec-2−/− vasculature during midgestation. (A-B) Confocal images of diencephalon sections with an antibody to phospho-Syk (Y519/520) (A) or an antibody to phospho-PLCγ2 (Y1217; arrows) (B). 4′,6-Diamidino-2-phenylindole is the nuclear stain. (C-D) Pseudocolored TEM of eMks (C) or platelets (D) in the PNVP vessel lumen of the lower diencephalon. eMk demarcation membrane system (asterisks), microparticles (red arrows), and lamellipodia (pink arrows) are indicated (C). Platelet filopodia projections (orange arrow) and granule secretion (red arrow) are indicated (D). (E) Confocal images of diencephalon sprouts and COL-1 expression. Dashed lines outline each sprout. (F) Confocal images of eMks with α-granules labeled with TSP-1 in the lower diencephalon. Insets show eMks in the midbrain. Bar graph (bottom) represents the percentage of fetal Mks with high or low TSP-1 threshold levels. (G) Confocal images of eMks isolated from E11.5 peripheral blood, that were either plated on a slide through cytospin as a negative activation control or incubated on a slide coated with COL-1 or PDPN for 30 minutes. The α-granules were labeled with TSP-1. The bar graph (right) represents eMk α-granule intensity. Data represent mean ± standard error of the mean and 3 experimental repeats with at least 3 technical repeats each. Scale bars, 10 μm (A-B); 2 μm (C); 500 nm (D); 20 μm (E-F); and 5 μm (G). **P < .01.
Further ultrastructural analysis indicated that, at E10.5, WT eMks around the diencephalon had an extensive demarcation membrane system and many granules, whereas in Pdpn−/− embryos they had a less developed demarcation membrane system, many lamellipodia processes, and many surrounding microparticles (Figure 4C) that were CD41+ (supplemental Figure 3F), suggesting activation.31 Consistently, WT platelets in the diencephalon vasculature were round and contained many granules, but in Pdpn−/− embryos, they had filopodia projections and apparent granule secretion (Figure 4D). These results are consistent with a heightened activation phenotype of eMks and platelets in the vasculature of Pdpn−/− and Clec-2−/− diencephalons at midgestation. Further RNA sequencing analysis of E10.5 brains revealed upregulated biological processes and cellular components related to platelet/eMk function and vascular development in Pdpn−/− vs WT brains (supplemental Figure 4A).
In addition to the PDPN-CLEC-2 signaling pathway, Syk can be activated through collagen signaling in Mks/platelets.24 The unexpected activation of the ITAM pathway in both Pdpn−/− and Clec-2−/− eMks led us to ask whether the ITAM pathway is activated through this alternative mechanism. To investigate, we stained embryos with an antibody to COL-4, which is a major component of the extracellular matrix of mature vasculature (supplemental Figures 3A and 4B). However, we detected low COL-4 levels (supplemental Figure 3A) and instead were surprised to find that COL-1 levels were expressed around angiogenic sprouts and were especially high around the PNVP at E10.5 (supplemental Figure 4C-D), but was not observed in the brain at E17.5. Collagen-1 induces platelet aggregation and secretion much more effectively than COL-4,32 and COL-1 levels were higher around aneurysm-like sprouts in Pdpn−/− and Clec-2−/− than in WT at E11.5 (Figure 4E).
To determine the activation status of eMks in vivo, we stained WT and mutant embryos with an antibody to thrombospondin 1 (TSP-1), an α-granule marker. Immunofluorescent imaging showed a considerable reduction in TSP-1 staining of eMks of Pdpn−/− and Clec-2−/− lower diencephalons relative to that of WT eMks (Figure 4F), although other brain regions showed similar levels, suggesting that there is increased granule secretion in mutant eMks in the lower diencephalon.
To test whether circulating eMks show differences in secretion in response to COL-1 or PDPN, we used an in vitro secretion assay of eMks isolated from E11.5 WT peripheral blood. There was a significant reduction in TSP-1 α-granule content in eMks on COL-1 plates, compared with eMks on either PDPN plates or negative controls (eMks cytospun onto slides; Figure 4G). These results support that there is a difference in eMk secretion in response to PDPN or COL-1 activation.
Increased TIE-2 signaling in sprout endothelial cells of Pdpn−/− and Clec-2−/− diencephalons
VEGF and ANGPT-1 are key angiogenic regulators. Confocal imaging revealed no difference of activated VEGF receptor VEGFR-233 on WT, Pdpn−/−, or Clec-2−/− angiogenic sprouts in the diencephalon (supplemental Figure 5A). However, the activated form of the ANGPT-1 endothelial receptor TIE-234 was significantly increased in both the Pdpn−/− and Clec-2−/− diencephalons relative to WT (Figure 5A). TIE-2 activation-mediated signaling counteracts VEGFR-2 signaling via sequestration of SRC, preventing phosphorylation and internalization of VE-cadherin.35 We found a significant decrease in activated, but not total, endothelial sprout SRC36 in Pdpn−/− and Clec-2−/− (Figure 5B; supplemental Figure 5B). VE-cadherin staining37 showed a slight increase in junctional staining at E10.5 (supplemental Figure 5C), and a significant increase on aneurysm-like sprouts by E11.5 in Pdpn−/− and Clec-2−/− (Figure 5C). These results support the notion that increased TIE-2 signaling in Pdpn−/− and Clec-2−/− angiogenic sprouts contributes to the growth of aneurysms until endothelial cells are stressed (Figure 5D) to the point of rupture.

Increased Tie2 signaling in sprout endothelial cells of Pdpn−/− and Clec-2−/− diencephalon. (A-B) Confocal images of diencephalon radial sprouts with an antibody to phospho-TIE-2 (Y991), a marker for TIE-2 activation (A), or antibody probe to phospho-SRC (Y418), a marker for SRC activation (B). Bar graphs represent the quantified ratio of sprout phospho-TIE-2 intensity (A) or phospho-SRC intensity compared with the paired WT (B). Dashed outlines show each sprout. (C-D) Confocal microscopy of junctional VE-cadherin expression in radial sprouts of diencephalon at E11.5 costained with anti-CD31, an endothelial cell junction marker (C), or with anti-ERG, a marker for endothelial cell nuclei (D). Bar graph on right represents the quantified ratio of sprout VE-cadherin intensity compared with paired WT (C). Data represent mean ± standard error of the mean; n, number of embryos. At least 3 sprouts/embryo analyzed. Scale bars, 20 μm. *P < .05; **P < .01.
Increased Tie2 signaling in sprout endothelial cells of Pdpn−/− and Clec-2−/− diencephalon. (A-B) Confocal images of diencephalon radial sprouts with an antibody to phospho-TIE-2 (Y991), a marker for TIE-2 activation (A), or antibody probe to phospho-SRC (Y418), a marker for SRC activation (B). Bar graphs represent the quantified ratio of sprout phospho-TIE-2 intensity (A) or phospho-SRC intensity compared with the paired WT (B). Dashed outlines show each sprout. (C-D) Confocal microscopy of junctional VE-cadherin expression in radial sprouts of diencephalon at E11.5 costained with anti-CD31, an endothelial cell junction marker (C), or with anti-ERG, a marker for endothelial cell nuclei (D). Bar graph on right represents the quantified ratio of sprout VE-cadherin intensity compared with paired WT (C). Data represent mean ± standard error of the mean; n, number of embryos. At least 3 sprouts/embryo analyzed. Scale bars, 20 μm. *P < .05; **P < .01.
PDPN reduces COL-1–induced ANGPT-1 secretion by fetal Mks
ANGPT-1 is the main activating ligand for TIE-2 and is primarily expressed in perivascular cells such as pericytes,38 but E10.5 angiogenic sprouts had few associated pericytes (supplemental Figure 1E). To determine whether eMks were a major source of ANGPT-1, we used in situ hybridization with the RNAscope probe to Angpt1, because we found no reliable antibody to ANGPT-1 for immunostaining on neural tissue sections. The result indicated that CD41+ eMks/platelets expressed high levels of Angpt1 at E10.5 (Figure 6A). To determine how different ligands effect ANGPT-1 release from Mks, we incubated isolated fetal Mks on plates coated with PDPN, COL-1, or COL-1 + PDPN (Figure 6B). After a 30-minute incubation, ANGPT-1 levels were significantly reduced in Mks on COL-1 plates in comparison with ANGPT-1 content in Mks on both PDPN and COL-1 + PDPN–coated plates, which remained unchanged (Figure 6C; supplemental Figure 6A), with levels similar to those of negative control plates coated with IgG Fc (Fc; supplemental Figure 6C). Incubation with Clec-2−/− Mks confirmed that secretion differences among the PDPN-coated plates were CLEC-2 dependent (Figure 6D; supplemental Figure 6B), indicating a specific inhibitory role for PDPN and CLEC-2 in the regulation of Mk secretion. Unc13d−/− fetal Mks, which lacked secretion, showed reduced ANGPT-1 secretion in the Mks on COL-1–coated plates (Figure 6E), indicating that WT fetal Mk differences are secretion dependent. ANGPT-1 and TSP-1 are both α-granule components.16,39 However, colocalization results showed that ANGPT-1 and TSP-1 were not in the same α-granules (supplemental Figure 6D), and instead ANGPT-1 colocalized more with vWF-containing α-granules. We also found a significant difference in TSP-1 levels of eMks between PDPN- and COL-1–coated plates (supplemental Figure 6E-F), supporting that PDPN-CLEC-2 regulates secretion of different cargo-containing α-granules. There appeared to be no secretion of TSP-1 or ANGPT-1 in adult bone marrow Mks after the cells were incubated on different ligand(s)-coated plates (supplemental Figure 6G). Adult peripheral blood platelets were also analyzed as a comparison. We found that platelets on either PDPN-Fc- or COL-1–coated plates had similarly increased secretion of TSP-1 compared with platelets on the plate coated with both PDPN-Fc and COL-1 (supplemental Figure 6H). This interesting result, the significance of which remains to be further investigated, suggests that adult platelets are more susceptible to activation in tissues where either PDPN or COL-1 is expressed.

PDPN reduces COL-1–induced ANGPT-1 secretion from fetal Mks. (A) RNAscope confocal images of diencephalon Angpt1 expression. Shown is colocalization with CD41+ cells (left; pink arrows) in the lower diencephalon. High-magnification images (right) show CD41+ cells colocalized with Angpt1 in the diencephalon neural tissue and the PNVP. (B) Overview of eMk isolation, culture, and static adhesion assay on recombinant PDPN/human IgG Fc chimera protein (PDPN-Fc), COL-1, or both. Plate coated with IgG Fc used as negative control (Fc). (C-E) Confocal images of ANGPT-1 levels of WT (C) or Unc13d−/− (E) eMks after incubation for 30 minutes on PDPN, COL-1, or both, or with Clec-2−/− eMk after incubation for 30 minutes on isotype IgG Fc, COL-1, or COL-1+PDPN (D), with comparison with WT. Bar graphs (right) represents the percentage of Mks with high, medium, or low ANGPT-1 levels. Data represent mean ± standard error of the mean; n, number of experimental repeats, with 3 technical repeats each. Scale bars, 50 μm (A, left); 20 μm (A, right); and 10 μm (C-E). **P < .01.
PDPN reduces COL-1–induced ANGPT-1 secretion from fetal Mks. (A) RNAscope confocal images of diencephalon Angpt1 expression. Shown is colocalization with CD41+ cells (left; pink arrows) in the lower diencephalon. High-magnification images (right) show CD41+ cells colocalized with Angpt1 in the diencephalon neural tissue and the PNVP. (B) Overview of eMk isolation, culture, and static adhesion assay on recombinant PDPN/human IgG Fc chimera protein (PDPN-Fc), COL-1, or both. Plate coated with IgG Fc used as negative control (Fc). (C-E) Confocal images of ANGPT-1 levels of WT (C) or Unc13d−/− (E) eMks after incubation for 30 minutes on PDPN, COL-1, or both, or with Clec-2−/− eMk after incubation for 30 minutes on isotype IgG Fc, COL-1, or COL-1+PDPN (D), with comparison with WT. Bar graphs (right) represents the percentage of Mks with high, medium, or low ANGPT-1 levels. Data represent mean ± standard error of the mean; n, number of experimental repeats, with 3 technical repeats each. Scale bars, 50 μm (A, left); 20 μm (A, right); and 10 μm (C-E). **P < .01.
Platelet aggregation and secretion are not required for vascular development during midgestation
Aggregation is a key platelet function. To determine whether it is critical for vascular development, we analyzed mice lacking platelet aggregation (deficiency of integrin β3 [β3−/−])40 and a mouse line lacking both platelet aggregation and secretion (β3−/−;Unc13d−/−; supplemental Figure 7A). No bleeding or vascular defects were found in β3−/− or β3−/−;Unc13d−/− embryonic brains relative to WT at E12.5 and E13.5 (supplemental Figure 7A-D), when there was 100% bleeding penetrance in the Pdpn−/− and Clec-2−/− embryos. These results indicate that platelet aggregation, Unc13d-regulated secretion, or both are not critical for brain angiogenic sprout development.
Among published mouse genetic models with functional deficiencies of Mks and platelets, we found that mice lacking Ras-GTPase activating protein RASA3, which is a critical negative regulator of platelet activation, present at E15.5 with hemorrhages that are similar to the Clec-2−/− phenotype.41 We thus asked whether PDPN-CLEC-2 regulates activity through RASA3 in eMks. However, E12.5 Rasa3−/− embryos had only 1 of 3 with a minor brain bleed and no change in vasculature development, although there was a reduced number of localized platelets and eMks around the diencephalon compared with WT (supplemental Figure 7E-H). These data suggest that the RASA3 pathway is not critical to brain angiogenic development at midgestation.
Blocking platelet activation mitigates Pdpn−/− vascular defects
Platelet inhibitors such as aspirin and ticagrelor (blocker of the adenosine 5′-diphosphate [ADP] receptor P2Y12) are commonly used to reduce platelet activation.42,43 To determine whether activation of eMks/platelets is necessary for defective angiogenesis in mice lacking PDPN, we treated pregnant dams with both aspirin and ticagrelor from E8.5 to 12.5 (Figure 7A). Analysis of platelets from treated pregnant dams before harvest confirmed defective aggregation (Figure 7B). The treatment resulted in an almost complete prevention of bleeding phenotypes within the diencephalon (Figure 7C). Analysis of vascular development showed normalized angiogenic sprouting in the Pdpn−/− diencephalon with treatment (Figure 7D-E). The number of eMks and platelets localized around the diencephalon was also significantly reduced compared with the vehicle-treated Pdpn−/− (supplemental Figure 8C-D). Treatment with ticagrelor or aspirin alone did not mitigate the phenotype (supplemental Figure 8A-B). In addition, treatment of cultured eMks with aspirin and ticagrelor in combination prevented secretion differences between PDPN- and COL-1–coated plates (Figure 7F). These results indicate that heightened activation of eMks and platelets are the primary cause of diencephalon hemorrhaging in the absence of PDPN.

Blocking platelet activation mitigates Pdpn−/− vascular defects. (A) Overview of strategy to block platelet activation: the pregnant dam was treated once daily (green arrows) by oral gavage, and embryos were harvested at E12.5 (blue arrow). (B) Platelet aggregation analysis from treated pregnant dams before embryo harvest. (C) Gross images of embryo heads, showing hemorrhages (arrows). Numbers represent embryos with bleeding (red)/total embryos (white). (D) Confocal images of forebrain vasculature and bleed areas (arrows). IB4, vessel; TER119, red cells. (E) Bar graph represents quantification of all diencephalon vascular sprouts from PNVP; n, number of embryos. (F) Bar graphs represent percentage of eMks with high, medium, or low ANGPT-1 threshold levels after a 30-minute treatment with either vehicle or 30 μmol aspirin + 10 μmol ticagrelor and then a 30-minute static adhesion on plates containing PDPN, COL-1, or both. Below are corresponding confocal images of representative eMks. Data represent means ± standard error of the mean; n, number of experimental repeats, with 3 technical repeats each. Scale bars, 500 μm (D) and 10 μm (F). **P < .01.
Blocking platelet activation mitigates Pdpn−/− vascular defects. (A) Overview of strategy to block platelet activation: the pregnant dam was treated once daily (green arrows) by oral gavage, and embryos were harvested at E12.5 (blue arrow). (B) Platelet aggregation analysis from treated pregnant dams before embryo harvest. (C) Gross images of embryo heads, showing hemorrhages (arrows). Numbers represent embryos with bleeding (red)/total embryos (white). (D) Confocal images of forebrain vasculature and bleed areas (arrows). IB4, vessel; TER119, red cells. (E) Bar graph represents quantification of all diencephalon vascular sprouts from PNVP; n, number of embryos. (F) Bar graphs represent percentage of eMks with high, medium, or low ANGPT-1 threshold levels after a 30-minute treatment with either vehicle or 30 μmol aspirin + 10 μmol ticagrelor and then a 30-minute static adhesion on plates containing PDPN, COL-1, or both. Below are corresponding confocal images of representative eMks. Data represent means ± standard error of the mean; n, number of experimental repeats, with 3 technical repeats each. Scale bars, 500 μm (D) and 10 μm (F). **P < .01.
Discussion
In mammals, the development of the brain coincides with rapid vascularization to meet its metabolic demands and deliver neurotrophic factors for growth. Vascularization of the brain primarily occurs through angiogenic sprouting of radial vessels from the PNVP during midgestation, after a defined spatial order (hindbrain > telencephalon > diencephalon). In both Pdpn−/− and Clec-2−/− embryos, aneurysms and hemorrhages primarily occur in the diencephalon, which develops radial angiogenic sprouts half a day later than the rest of the developing brain at E10.44,45 This time point is concomitant with establishment of fully functional circulation in mice,46 suggesting that hemodynamic pressure contributes to susceptibility in the formation of aneurysms within this region.47
In adult humans and mice, the roles of Mks and platelets derived from HSCs have been well studied.48 However, Mks are generated differently during early embryonic gestation.1 Megakaryopoiesis is an important component of primitive hematopoiesis, which begins to develop in the yolk sac at ∼E7.5 in mice.49 During midgestation, these low ploidy CD41+ cells would primarily be Mks from the first primitive wave of hematopoiesis (Figure 2G).5 It is of note that erythromyeloid progenitor (EMP)–derived Mk progenitors from the yolk sac emerge soon after primitive hematopoiesis begins and also produce Mks during this period of development. Therefore, even though most Mks that we analyzed from E10.5 to E11.5 were “primitive,” some may be EMP-derived Mks, as there are no markers to distinguish both. Therefore, we chose to use the term embryonic Mks. eMks are in the circulation beginning at ∼E10 and have been described as diploid platelet-forming cells.50 These cells differ from definitive Mks in kinetics of growth and cytokine sensitivity.6 In addition, we found that circulating eMks have ectoplasmic lamellipodia processes that resemble nonmammalian vertebrate diploid thrombocytes,28 which have less efficient hemostatic functions than mammalian adult platelets.51 During gestation, embryonic circulation is highly thrombogenic, partially because of higher levels of ultralarge multimer von Willebrand factor, in comparison with adult circulation.52-54 Our results also revealed that neural vascular matrix contains COL-1, which is much more thrombogenic than the COL-4 commonly found in the subendothelial matrix of adult tissues.32 Therefore, diploid thrombocyte-like eMks are presumably important in maintaining hemostasis in the hypercoagulant embryonic circulation.
Platelet ITAM signaling is critical for vascular integrity during inflammation in adult mice.21,24 However, whether Mks and platelets are important for nascent vascular integrity at midgestation remains unclear. Mice lacking the transcription factor p45NF-E2 have severe thrombocytopenia but show no signs of embryonic brain hemorrhage.55,56 On the other hand, mice lacking the transcription factor Meis-1 lack Mks and present at midgestation with brain hemorrhaging,57,58 indicating a critical function for eMks. In addition, mice lacking the transcription factor Runx-1 exhibit reduced eMks by E10.5 and present at midgestation with hemorrhaging phenotypes similar to those of PDPN and CLEC-2 knockout mice, suggesting a critical role for eMks during development.50,59
PDPN binds to CLEC-2 activating hemITAM signaling through the phosphorylation of Syk and downstream activation of PLCγ2, leading to platelet aggregation. However, mice lacking Syk have a milder brain-bleeding phenotype than those lacking CLEC-2,20,60 whereas mice lacking PLCγ2 do not show any obvious brain bleeding at midgestation.61 Our extensive analysis revealed no vascular defects or bleeding in β3−/− embryonic brains by E13.5. Collagen also activates Syk through interaction with its receptor GPVI and FcRγ2, but mice lacking either GPVI or FcRγ2 are viable with no spontaneous brain bleeding.62,63 Loss of genes critical to platelet activation, such as CalDAG-GEFI,64 RAP1a/b,65 and Gα2,66 do not show any obvious brain bleeding phenotype, but loss of critical Ras GTPase-activating protein-3 (RASA-3) results in severe hemorrhaging by E15.5, and double knockout with CalDAG-GEFI rescues the Rasa3−/− phenotype,41 suggesting that regulation of platelet activity is critical to embryonic development.
During midgestation, the diencephalon expresses high levels of PDPN and COL-1. Our data showed that PDPN counteracted the action of COL-1, preventing secretion of granule content from eMks, including ANGPT-1, the main activating ligand for TIE-2. Constitutively activated TIE-2 is known to contribute to congenital vascular abnormalities.13 Mutations of TIE-2 resulting in hyperphosphorylation cause venous malformation in patients.67 Increased VE-cadherin is found in early saccular aneurysms.68 The aneurysm-like sprouts in Pdpn−/− and Clec-2−/− embryos have increased TIE-2 activity and increased junctional VE-cadherin intensity,35 supporting a possibility that increased junctional VE-cadherin leads to growth of aneurysms until they reach a size where increased stress results in rupture around E11.5, when hemodynamic pressures increase in the circulation.69 These results reveal new insights into the differential function of PDPN and CLEC-2 in regulation of eMk/platelet activation during early vascular development in the brain.
ADP and thromboxane A2 (TXA2) are necessary for tyrosine phosphorylation response upon CLEC-2 activation.70 Interestingly, we found that aspirin, which blocks thromboxane A2 action, and ticagrelor, an ADP receptor P2Y12 blocker, in combination, but not individually, mitigated vascular defects and bleeding phenotypes of Pdpn−/− embryos. This finding highlights the complexity of the CLEC-2 signaling cascade. It also suggests that dual antiplatelet therapy may be useful for treating congenital vascular abnormalities with risks of bleeding, such as saccular aneurysms with enhanced TIE-2 activation.71
The spontaneous hemorrhages in Pdpn−/− or Clec-2−/− embryonic brain resembles germinal matrix-intraventricular hemorrhage (GMH-IVH), which affects ∼35% of premature infants, leading to high mortality or subsequent disabilities.72 Impaired vascular stability is a key pathological feature of GMH-IVH. Of interest, the nonsteroidal agent indomethacin inhibits platelet function and has been shown in clinical trials to reduce grade 3 and 4 GM-IVH when given prophylactically in very low birth weight infants.73 Although the etiology of GMH-IVH is likely multifactorial and complex, it is plausible that impaired PDPN and/or CLEC-2 function contributes to the pathogenesis of a subset of this common human disease. In addition, mice lacking PDPN or CLEC-2 with fully penetrant central nervous system bleeding phenotypes may serve as valuable animal models for exploring new mechanistic insights into the pathogenesis of GMH-IVH and for testing potential new therapies.
For publication-related data, please e-mail the corresponding author, Lijun Xia (lijun-xia@omrf.org).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank James George for critical comments, and the Imaging Core Facility of the Oklahoma Medical Research Foundation for tissue processing and electron microscopy.
The study is supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grants HL128390, HL149860, HL153728 (L.X.), HL134210 (C.H.), and HL133668 (W.B.); NIH, National Institute of Child Health and Human Development grant HD083418 (L.X.); and NIH, National Institute of General Medical Sciences grant GM114731 (R.P.M.).
Authorship
Contribution: C.H., Y.K., B.S., M.J.M., R.L., and S.M. performed the experiments; C.H., Y.K., B.S., R.P.M., and L.X. designed the research and analyzed the data; S.W. and W.B. provided reagents and comments; and C.H. and L.X. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lijun Xia, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: lijun-xia@omrf.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal