


Abstract
Approximately 80% of adult patients with immune thrombocytopenia (ITP) have treatment failure with corticosteroids or become dependent on them and require second-line therapy. Several new and effective therapies have been introduced during the past decade and our understanding of disease burden and its effect on quality of life has expanded. It is now recommended that splenectomy, the standard second-line therapy for decades, be delayed for at least 12 to 24 months, allowing for more patients to achieve remission on medical therapies before considering surgery. It is highly recommended that medical therapies be used that have abundant clinical trial evidence, such as the thrombopoietin receptor agonists (TPO-RAs) rituximab and fostamatinib. Unfortunately, there are no reliable biomarkers that help in treatment selection. These therapeutic medical options have variable efficacy, safety profiles, mechanisms of action, and modes of administration. This enables and mandates an individualized approach to treatment, where patient involvement, preferences and values have become central to the process of choosing the appropriate therapy. Both TPO-RAs and fostamatinib are maintenance therapies, whereas rituximab is given for a limited number of doses. Although the response is usually maintained while receiving a TPO-RA or fostamatinib therapy, half of rituximab responders will no longer respond 1 to 2 years after administration and require retreatment or other therapy.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune disease affecting 2 to 4 per 100 000 individuals annually with an overall prevalence of ∼10 per 100 000 individuals.1,2 ITP is characterized by an isolated platelet count <100 × 103/μL, which increases the bleeding risk.3 Diagnosis of primary ITP relies on exclusion of nonimmune causes of thrombocytopenia (myelodysplastic syndrome and hereditary thrombocytopenia) and of other conditions that can cause secondary immune-mediated thrombocytopenia such as autoimmune diseases (systemic lupus erythematosus), malignancies (chronic lymphocytic leukemia), infections (hepatitis C virus and HIV) and drugs.4 Differentiation between primary and secondary ITP is important, because the focus of management of secondary ITP is usually to treat the primary cause.
The pathophysiology of ITP is complex and still not completely understood. The traditional basic concepts involve antibody-mediated platelet destruction, predominantly in the spleen and liver via Fcγ receptors,5 and impaired megakaryocyte platelet production.6 Direct T-cell toxicity to platelets and megakaryocytes has more recently been shown to be an important mechanism.7
Clinical manifestations of ITP range from totally asymptomatic to increased bruising and petechiae, and rarely, to major or fatal bleeding. ITP treatment attempts to reduce platelet and megakaryocyte destruction through immune modulation and splenectomy or to increase platelet production by megakaryocytes with thrombopoietin receptor agonists (TPO-RAs).8-11 Corticosteroids are the first-line treatment. Although >75% of adult patients respond to corticosteroids, only 20% to 30% achieve sustained remission upon discontinuation.12 Although corticosteroid-responsive patients may maintain an adequate platelet count indefinitely with corticosteroid continuation, the adverse effects are usually unacceptable and most corticosteroid-dependent patients eventually require subsequent treatment. Treatment decisions depend on various factors related to the patient, the disease, and the health care system. Updated ITP clinical guidance documents can help make these decisions.8-11 The aim of this article is to provide insight on personalizing therapy in adult patients with ITP who are unresponsive to or dependent on corticosteroids, using information from the new clinical guidance documents and the experience of the authors. The number of patients unresponsive to or dependent on corticosteroids may be reduced in the future by more aggressive initial treatments, such as adding recombinant TPO13 or rituximab,14,15 but those treatments have not yet been incorporated into any clinical guidance documents or into our practice.
Whom do we treat?
There are 3 reasons for treating ITP.
Preventing bleeding: although only 5% of patients present with severe bleeding,16 the bleeding leads to hospital admission within 5 years after diagnosis in ∼15%.17 Therefore, stopping ongoing bleeding and preventing future serious bleeding episodes are the prime treatment goals. Prevention requires assessment of bleeding risk, but no bleeding scores have been validated in adults to assess bleeding risk,18-20 and therefore assessment still relies on platelet counts and clinical evaluation (age, previous bleeding events, comorbidities, concomitant medications, and activity level). In line with current clinical guidance documents, we usually reserve therapy for patients with platelets <30 × 103/μL or <50 × 103/μL in patients with increased bleeding risk related to irreversible causes (eg, intestinal angiodysplasia, spontaneous intracranial bleeding, frequent falls, or anticoagulant therapy).8,9 Conversely, we lowered the threshold to <20 × 103/μL in totally asymptomatic patients or those in whom treatment options were exhausted.
Remission induction: remission is poorly defined in ITP but may be regarded as either normalization of the platelet count (“complete remission”) or a sustained elevation of platelet count to a hemostatic level (“treatment-free partial remission”) without the need for continued therapy.
Improving quality of life and alleviating symptoms: ITP affects various aspects of health-related quality of life (HRQoL). Randomized controlled trials (RCTs) have demonstrated improvements in various HRQoL dimensions for treatment with a TPO-RA compared with a placebo or standard of care.21,22 Generally, elevating platelet count by any therapy has been shown to improve HRQoL.23 Although patients with ITP frequently complain of fatigue correlated with a decrease in platelet count, it is our experience that fatigue does not always improve with an increase in platelet count.24
Our patients
The following 5 vignettes illustrate how we personalize therapy for adult patients with ITP who are unresponsive to or dependent on corticosteroids.
Patient 1: young female
A 23-year-old woman presented with generalized mucocutaneous bleeding and platelet count of 3 × 103/μL. She had no other symptoms, and the physical examination was negative except for large bruises and petechiae over the lower extremities and on the buccal mucosa. A diagnosis of ITP was made, and she was treated with dexamethasone 40 mg/d for 4 days. Her platelet count began increasing by day 4, reaching 112 × 103/μL at day 14 but then declining by day 25 to 11 × 103/μL. Because she responded to the first cycle of dexamethasone and treatment was well tolerated, a second cycle was initiated. After a second response the patient again relapsed. At a follow-up visit 10 weeks after diagnosis, she had a platelet count of 9 × 103/μL, cutaneous bruises, and heavy menstrual bleeding. She noted increasing fatigue and anxiety, fearing the consequences of disease. She worried whether she would ever be able to undergo pregnancy safely.
Patient 2: elderly patient
A 78-year-old woman was referred because of an abnormal complete blood count found during a routine check-up: platelet count 12 × 103/μL (215 × 103/μL 6 months earlier), hemoglobin 12.8 g/dL, white blood cell (WBC) count 5.2 × 103/μL (normal differential), and blood smear remarkable only for the decreased number of platelets. She had hypertension, diabetes, and osteoporosis and was taking aspirin. She had bruises on her hands. Aspirin was discontinued, and prednisone was initiated at 1 mg/kg per day. Platelet count increased to 62 × 103/μL after 2 weeks. Prednisone was gradually tapered by 5 to 10 mg/d weekly, but the platelet count fell to pretreatment levels at 10 mg/d, prompting a return of prednisone dose to 30 mg/d. Her blood sugar became persistently elevated requiring addition of another antidiabetic agent. At a follow-up visit 12 weeks after diagnosis, she was still receiving prednisone 20 mg/d with a platelet count of 34 × 103/μL.
Patient 3: complicated patient receiving anticoagulation
A 69-year-old man presented with platelets of 10 × 103/μL, generalized mucocutaneous bleeding, and recurrent epistaxis. Hemoglobin was 12.2 g/dL, WBC 3.1 × 103/μL, and mean corpuscular volume 78 fL, with a normal blood smear, except for reduced platelets. He had atrial fibrillation and hypertension and took apixaban 5 mg twice daily in addition to antihypertensive medication. Apart from thrombocytopenia, a diagnostic work-up revealed only ITP along with mild iron deficiency. Apixaban was immediately discontinued. Given his age and the uncertainty of whether he would tolerate dexamethasone, prednisone 60 mg daily was initiated. Two weeks later, his platelet count was still 10 × 103/μL, but he had no bleeding.
Patient 4: patient needing elective surgery
A 39-year-old man was referred for a presurgical evaluation before an elective L4 to L5 laminectomy with a platelet count of 27 × 103/μL. Three years earlier, a platelet count of 22 × 103/μL was noted on a routine physical examination, with no bleeding and no other potential causes. At that time, he was given prednisone 60 mg/d, and within 8 days, his platelet count rose to 46 × 103/μL, but his blood sugar rose to 350 mg/dL. Prednisone was discontinued. Since then, he has been monitored with no therapy, with platelets 25 × 103/μL to 38 × 103/μL. He has no symptoms or other medical problems except for obesity (body mass index, 35).
Patient 5: patient seeking a cure
An otherwise healthy 46-year-old airline pilot presented with easy bruising, petechiae, and a platelet count of 12 × 103/μL. He had Hashimoto thyroiditis and took only thyroid replacement medication. Three years ago, he had had a platelet count of 6 × 103/μL and hematuria and had been treated with prednisone 60 mg/d with his platelet count rising to 181 × 103/μL. Corticosteroids were tapered over 9 months, and his platelet count remained at 105 × 103 to 135 × 103/μL. He was frustrated by the recurrence of ITP because he had been told the corticosteroids had “cured” his disease. He was fearful that he would lose his certification as a commercial pilot.
How do we choose treatment?
The recent clinical guidance documents from the American Society of Hematology (ASH) and the International Consensus group do not express preference for initial corticosteroid type, but strongly recommend limiting corticosteroid treatment for 6 to 8 weeks maximum and quickly switching to other therapy if unresponsive or corticosteroid dependent.8,9 Although based on a low level of evidence, all recent clinical guidelines on ITP advocate early (>3 months from diagnosis) use of second-line treatments such as a TPO-RA or rituximab to avoid corticosteroid toxicity.
Second-line treatment is ultimately required in 70% to 80% of steroid-treated patients. Although splenectomy has historically been used in this setting, patient preference and widespread availability of effective medical therapies has reduced its use.25 There are many second-line therapeutic options, but few have been evaluated in RCTs and include TPO-RAs (romiplostim, eltrombopag, and avatrombopag), rituximab, and fostamatinib (Table 1). The remaining therapeutic options have been evaluated mostly in single-arm prospective or retrospective studies and have included several classes of immune-suppressive/modulatory agents all with varying responses, including azathioprine, cyclophosphamide, cyclosporine, mycophenolate mofetil, danazol, dapsone, and vinca alkaloids.8 However, these drugs will not be discussed in this review.
Therapies with adequate RCT evidence
| Therapy | Route of administration | Mechanism of action | Dosage | Response rate | Adverse effects |
|---|---|---|---|---|---|
| Romiplostim | Subcutaneous injections | TPO-RA | 1-10 µg/kg once weekly | Overall response of 75%; durable response at 6 mo is 65%; 10% to 30% may achieve treatment remission | Headache, muscle aches, venous and arterial thromboembolism, possible increase in bone marrow reticulin, and collagen fibrosis |
| Eltrombopag | Oral (restricted diet) | TPO-RA | 25-75 mg once daily | Headache, venous and arterial thromboembolism, elevated liver enzymes, possible increase in bone marrow reticulin, and collagen fibrosis | |
| Avatrombopag | Oral | TPO-RA | 20-40 mg once daily | 65% at day 8* | Headaches, arthralgia, and venous and arterial thromboembolism |
| Rituximab | Intravenous administration | Immunosuppressive; anti-CD20 | Infusions of 375 mg/m2 each week for 4 weeks or 1000 mg every other week, for 2 weeks† | Initial response rate 60%; durable response rate at 6-12 mo is ∼40% and at 5 y is ∼20% to 30% | Infusion-related side effects (chills, upper respiratory discomfort, and bronchospasm), neutropenia, hypogammaglobulinemia, serum sickness, increased risks of infection and progressive multifocal leukoencephalopathy (very rare). |
| Fostamatinib | Oral | Immunosuppressive; splenic tyrosine kinase inhibitor | 100-150 mg twice daily | Overall response 43%‡; stable response 18%§ | Hypertension, diarrhea, nausea, and transaminitis |
| Therapy | Route of administration | Mechanism of action | Dosage | Response rate | Adverse effects |
|---|---|---|---|---|---|
| Romiplostim | Subcutaneous injections | TPO-RA | 1-10 µg/kg once weekly | Overall response of 75%; durable response at 6 mo is 65%; 10% to 30% may achieve treatment remission | Headache, muscle aches, venous and arterial thromboembolism, possible increase in bone marrow reticulin, and collagen fibrosis |
| Eltrombopag | Oral (restricted diet) | TPO-RA | 25-75 mg once daily | Headache, venous and arterial thromboembolism, elevated liver enzymes, possible increase in bone marrow reticulin, and collagen fibrosis | |
| Avatrombopag | Oral | TPO-RA | 20-40 mg once daily | 65% at day 8* | Headaches, arthralgia, and venous and arterial thromboembolism |
| Rituximab | Intravenous administration | Immunosuppressive; anti-CD20 | Infusions of 375 mg/m2 each week for 4 weeks or 1000 mg every other week, for 2 weeks† | Initial response rate 60%; durable response rate at 6-12 mo is ∼40% and at 5 y is ∼20% to 30% | Infusion-related side effects (chills, upper respiratory discomfort, and bronchospasm), neutropenia, hypogammaglobulinemia, serum sickness, increased risks of infection and progressive multifocal leukoencephalopathy (very rare). |
| Fostamatinib | Oral | Immunosuppressive; splenic tyrosine kinase inhibitor | 100-150 mg twice daily | Overall response 43%‡; stable response 18%§ | Hypertension, diarrhea, nausea, and transaminitis |
Platelet counts >50 × 103//μL.
Lower doses down to 100 mg have been used.
One or more platelet counts ≥50 × 103/μL during weeks 0 to 12 of the study.
Platelet counts ≥50 × 103/μL at ≥4 of 6 biweekly clinic visits during weeks 14 to 24 of the study.
Current clinical guidelines encourage individualizing therapy and shared decision making, based on assessment of the patient’s medical condition, duration and severity of disease, and the patient’s values and preferences. Although “patient values and preferences” were not defined in any of the clinical guidelines, a suggested list is provided in Table 2. Based on these individual values and preferences, the appropriate therapy is chosen.
Patient values and preferences
| Fear and anxiety about low platelet count |
| Acceptance of a low platelet count |
| Tolerance of minor bleeding |
| Pregnancy planning |
| Activity level |
| Acceptance of chronic therapy vs limited administrations |
| Preference of daily tablets or weekly injections |
| Desire to avoid corticosteroids |
| Desire to avoid splenectomy |
| Desire to “get it all over with” |
| Cosmetic embarrassment over bruises and petechiae |
| Fear and anxiety about low platelet count |
| Acceptance of a low platelet count |
| Tolerance of minor bleeding |
| Pregnancy planning |
| Activity level |
| Acceptance of chronic therapy vs limited administrations |
| Preference of daily tablets or weekly injections |
| Desire to avoid corticosteroids |
| Desire to avoid splenectomy |
| Desire to “get it all over with” |
| Cosmetic embarrassment over bruises and petechiae |
These values and preferences may change considerably from the time of initial diagnosis to later persistent and chronic stages of the disease.
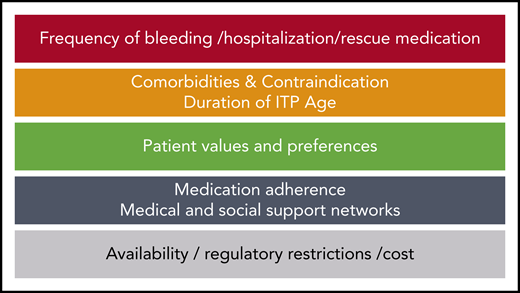
Our general approach (Figure 1) is as follows:
Assess how quickly we need to elevate the platelet count. Patients with bleeding or at high risk of bleeding, require therapies such as a TPO-RA or fostamatinib that rapidly elevate platelet count (1-2 weeks). Some patients may require brief reinstitution of higher doses of corticosteroids or IV immunoglobulin (IVIG) to regain control of the disease.
Evaluate factors related to the patient (comorbidities, age, concomitant medications, and activity level) and ITP (duration and severity) that influence treatment choice. For example, splenectomy is not recommended for the first 1 to 2 years after diagnosis or in patients with comorbidities that may impose high operative and/or postoperative risk.8,9,26 Patients aged >60 to 70 years have a reduced response to splenectomy.26 Rituximab may be more effective in younger females with short disease duration.27,28
Discuss with the patient (and often the family) the relevant facts regarding response rate, potential for remission, and safety and mode of administration of the potential therapeutic choices. Discuss platelet count goal; patients should be told that we do not seek a normal platelet count but simply one that mitigates bleeding and is “safe.” Some patients do quite well with no therapy at platelet counts as low as 15 × 103/μL to 20 × 103/μL. Patient compliance may predict whether an injection or infusion is preferred over oral therapy.
Determine available options based on regulatory restrictions, availability, and insurance coverage and cost.
Characteristics of available therapies
TPO-RAs
Romiplostim (weekly subcutaneous injections), eltrombopag, and avatrombopag (daily oral medications) are equally effective medications, having an overall response rate of ∼75%29 and a durable response rate (response at 6 months) of ∼65%.9,30 Avatrombopag is a newer TPO-RA similar to eltrombopag, but with no food interactions (and is recommended to be taken with food) or need for follow up on liver enzymes. The 24-week phase 3 study showed that the median cumulative weeks with platelet count >50 × 103/μL was much higher in avatrombopag-treated than in placebo-treated patients (12.4 vs 0.0 weeks; P < .0001).31 Three of 33 patients (9.4%) developed thromboembolism during the RCT and another patient developed jugular vein thrombosis in the open-label extension phase. Moreover, headache was reported in 37%, although exposure-adjusted incidence was not higher than that in the placebo arm.31
Several reports have shown that 10% to 30% of patients may discontinue the TPO-RA without reducing the response.32,33 For those who do not respond to one TPO-RA, switching to another yields a response in 50% to 80% of patients.34-36 Generally, the effect of a TPO-RA is seen within 2 weeks. They appear to be safe. Initial concerns regarding potential oncogenic effect and bone marrow fibrosis have not been substantiated.37-40 However, there is a possible increased risk of venous and arterial thromboembolism.41 The real and potential adverse effects of romiplostim, eltrombopag, and avatrombopag have been extensively reviewed.30,42,43
Fostamatinib
Fostamatinib is another oral agent recently licensed in the United States. In 2 RCTs, a stable platelet response (defined as at least 4 of 6 biweekly platelet counts ≥50 × 103/µL, in absence of rescue, during weeks 14-24) was seen in 18% of fostamatinib-treated patients vs 2% of placebo-treated ones.44 However, overall fostamatinib response (at least 1 platelet count ≥50 × 103/µL in the first 12 weeks) was 43% vs 14% with placebo, with time to response being 2 weeks. This 24-week study was markedly affected by allowing for patients in both arms to enter the open-label phase of the study early; 84% of patients receiving placebo and 60% of those receiving fostamatinib left the randomized study after 12 weeks because of lack of response. Fostamatinib had side effects of diarrhea, hypertension, and nausea in 29%, 20%, and 19% of patients, respectively.44
Rituximab
Rituximab has been used for ITP for >20 years. Although up to 60% respond initially,9,45,46 durable response rate at 6 to 12 months is ∼40% and at 5 years is ∼20% to 30%.47-49 It has been suggested that females who are <40 years of age attain more long-term remissions (47% at 48 months; 47% at 72 months) than other patients (33% at 48 months; 25% at 72 months).28 However, this finding has been disputed.48 Retreatment of relapsed patients usually induces a new remission, but it is unlikely to be maintained beyond the initial response duration. Because rituximab may reactivate hepatitis B infection, all patients must be tested before administration. Necessary vaccinations should be administered before rituximab treatment, as patients are unlikely to have a vaccination response for at least 6 months after their last infusion.50 Apart from infusion-related reactions, transient clinically insignificant neutropenia, and hypogammaglobulinemia, rituximab is safe.47
Splenectomy
Splenectomy is highly effective (70% long-term remission rate), but because of complications, a largely unpredictable response, and availability of safer medical options, it has become a less favored treatment.51 It is currently recommended that splenectomy be avoided during the first 1 to 2 years after diagnosis.8,9 In those patients preferring early splenectomy, we advocate a course of rituximab before the procedure, as suggested in the American Society of Hematology Guidelines.9
Clinical research studies
Several experimental medications have shown a promising effect and have entered phase 2 and 3 clinical trials. These include neonatal FcR antagonists, Bruton kinase inhibitor, and complement inhibitors.4 Inclusion in clinical studies is highly recommended for eligible patients.
Our approach was occasionally modified based on the factors shown in Figure 1. Figure 2 shows our general approach to the choice of therapy.
![Treatment algorithm for steroid dependent/unresponsive patients. In those patients accepting splenectomy as the second-line therapy, a course of rituximab is recommended before surgery (making sure vaccinations are completed before the rituximab). Most patients who do not accept splenectomy will respond to TPO-RAs, and such treatment should not be regarded as indefinite. Those in whom the platelet count does not respond to a TPO-RA or to whom such agents are not available can be treated with another medical therapy (another TPO-RA [alt TPO-RA], if available, or rituximab or fostamatinib). If one agent in this group fails, it is reasonable to try a different one. Treatment with immunosuppressive/immunomodulatory agents (we favor danazol, dapsone, azathioprine, or mycophenolate mofetil) can be considered. In those in whom 2 or more second-line therapies fail, reassess the diagnosis and the need for therapy. Such patients may be considered for a clinical trial or splenectomy. Splenectomy is best avoided for 1 to 2 years after diagnosis, if possible, but may be preferred by some patients in whom second-line or subsequent therapies fail. Boxes outlined in dashed lines denote alternative steps in treatment algorithm. Although clinical trials are suggested for the more refractory patients in this algorithm, they are also appropriate for patients earlier in their disease course.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2021010968/1/m_bloodbld2021010968f2.png?Expires=1769568938&Signature=b0PbdoEOA9VcRCS0lTn0LGnj7FupkOy96TwvliYrJ5VlHcA7PoqgwsHHUKjzhENyEJcx8R8dpPJhWOkSNcXFDWQxzkRgY2mplEAKFDyKj78WgVzewGb69-2i5WDnQc8M1IcjYjL6r0SH1yhbxndsS-T1Xyokq3xhOgMzqVGJpT58z806X~CtEqWhLXcUhrUN4dz1ZdFAAx2dapxBZg7UE48xxBL3br5Et1s~sd6EjnAXUKh7w9L8TDJHxlNrlCGTx4XGdOjU3AqDaBHwAEj3AMgg1QCh7bJtq8gBa-MM0lDfSRuTdWO1ZOdXkwku5426Dwp4rTlIguta~K-fDsE0bQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Treatment algorithm for steroid dependent/unresponsive patients. In those patients accepting splenectomy as the second-line therapy, a course of rituximab is recommended before surgery (making sure vaccinations are completed before the rituximab). Most patients who do not accept splenectomy will respond to TPO-RAs, and such treatment should not be regarded as indefinite. Those in whom the platelet count does not respond to a TPO-RA or to whom such agents are not available can be treated with another medical therapy (another TPO-RA [alt TPO-RA], if available, or rituximab or fostamatinib). If one agent in this group fails, it is reasonable to try a different one. Treatment with immunosuppressive/immunomodulatory agents (we favor danazol, dapsone, azathioprine, or mycophenolate mofetil) can be considered. In those in whom 2 or more second-line therapies fail, reassess the diagnosis and the need for therapy. Such patients may be considered for a clinical trial or splenectomy. Splenectomy is best avoided for 1 to 2 years after diagnosis, if possible, but may be preferred by some patients in whom second-line or subsequent therapies fail. Boxes outlined in dashed lines denote alternative steps in treatment algorithm. Although clinical trials are suggested for the more refractory patients in this algorithm, they are also appropriate for patients earlier in their disease course.
Treatment algorithm for steroid dependent/unresponsive patients. In those patients accepting splenectomy as the second-line therapy, a course of rituximab is recommended before surgery (making sure vaccinations are completed before the rituximab). Most patients who do not accept splenectomy will respond to TPO-RAs, and such treatment should not be regarded as indefinite. Those in whom the platelet count does not respond to a TPO-RA or to whom such agents are not available can be treated with another medical therapy (another TPO-RA [alt TPO-RA], if available, or rituximab or fostamatinib). If one agent in this group fails, it is reasonable to try a different one. Treatment with immunosuppressive/immunomodulatory agents (we favor danazol, dapsone, azathioprine, or mycophenolate mofetil) can be considered. In those in whom 2 or more second-line therapies fail, reassess the diagnosis and the need for therapy. Such patients may be considered for a clinical trial or splenectomy. Splenectomy is best avoided for 1 to 2 years after diagnosis, if possible, but may be preferred by some patients in whom second-line or subsequent therapies fail. Boxes outlined in dashed lines denote alternative steps in treatment algorithm. Although clinical trials are suggested for the more refractory patients in this algorithm, they are also appropriate for patients earlier in their disease course.
How did we approach our patients?
Patient 1: young female
Current clinical guidelines recommend treating patients with platelet count <20 × 103/µL to 30 × 103/µL. Several studies have shown that serious bleeding risk is increased when platelet count is <20 × 103/µL. In addition to thrombocytopenia, our patient had anxiety and fatigue and was concerned regarding future pregnancies. With no life-threatening bleeding, there was no need for rapid platelet count elevation. Had her menstrual bleeding become more significant, the addition of hormonal contraception or tranexamic acid could have been considered. A TPO-RA, rituximab, or fostamatinib were possible options. Depending on when she was considering pregnancy, she may have wished to avoid TPO-RAs and fostamatinib. Because there was some evidence indicating that females <40 years of age may have a 73% response rate (56% complete response), rituximab may be the most appropriate choice.28 TPO-RAs are usually considered before rituximab, because they are well tolerated and very effective, and up to 30% of patients taper off therapy in the first year and maintain safe platelet counts.32 Although 78% of second-line patients responded to fostamatinib in the 2 phase 3 studies, only 50% had counts that were consistently >50 × 103/µL52 ; because of the gastrointestinal side effects, we would consider other medications first. After discussing treatment advantages and disadvantages and evaluating her values and preferences, rituximab was chosen. She received two 1000-mg rituximab infusions 2 weeks apart. After 4 weeks, her platelet count was 102 × 103/µL. This dosing regimen is as effective as 1 infusion of 375 mg/m2 per week for 4 weeks.53 Lower doses have been suggested, but may have yielded a briefer response.54 Although some recommend avoidance of pregnancy for 12 months after rituximab,55 that opinion has not been supported by subsequent evidence.56,57
Patient 2: elderly patient
Current clinical guidelines recommend limiting the maximum duration of corticosteroid treatment to 6 to 8 weeks.8,9 The patient had already been treated with corticosteroids for 3 months and still required a relatively high prednisone dose to maintain an adequate platelet count. Her diabetes worsened and required additional antidiabetic agents. We started tapering prednisone, having a plan ready for the next treatment, should the count decrease. The patient had a high bleeding risk because of advanced age, comorbidities, and aspirin use. Hence, her platelet count should not be allowed to decline to <30 × 103/µL. Choosing among the 3 medical therapies based on their durable response rates (at 6 months) favored a TPO-RA (∼65%) over rituximab (∼40%) and fostamatinib (18%).9,44 A possible increased thromboembolism risk with TPO-RAs should be considered. Because of its potential side effects and lower durable response rate, we would not consider fostamatinib at this stage. The therapeutic options were discussed with the patient, and she opted for a TPO-RA because of the high response rate and few side effects. Because she preferred an oral agent with no dietary restrictions, avatrombopag 20 mg/d was initiated instead of eltrombopag. Two weeks after initiation of avatrombopag therapy, her platelet count increased to 72 × 103/µL, and during the next 12 months, it fluctuated between 59 × 103/µL and 88 × 103/µL. Had she not responded to avatrombopag, had a reduced response, or experienced side effects, switching to another TPO-RA may have been effective, as studies have shown.34,35 Addition of low doses of corticosteroids (up to 5 mg/d of prednisone) may augment the response in patients who do not respond optimally to a TPO-RA.58 Given that the patient had a stable platelet count for 6 to 12 months, we considered tapering the TPO-RA dose. Most patients maintain a safe count (>50 × 103/µL) at a lower dose, thus reducing cost and potential toxicity. Avatrombopag was tapered (every other week) to 20 mg every other day, then to 20 mg every third day. At that dose, the platelet count declined to 30 × 103/µL, prompting a return to 20 mg every other day.
Patient 3: complicated patient receiving anticoagulation
Patients with ITP who are receiving anticoagulation represent a challenge for the hematologist. Although there is no evidence-based platelet count at which anticoagulation can be safely given, most experts agree on a platelet threshold of >50 × 103/µL.8 However, this threshold should be adjusted according to the underlying indication and the risk of bleeding (eg, the patient with recent massive venous thromboembolism should not discontinue anticoagulation for a prolonged period and therefore a more rapid elevation and lower platelet count are accepted in contrast to the patient with atrial fibrillation who may remain without anticoagulation longer). Although usually not needed, because of his failure to respond to corticosteroids and his anemia, a bone marrow examination was performed that excluded other causes of thrombocytopenia. In general, we recommend that patients who fail to respond to any initial therapy (corticosteroids, IVIG, and anti-D) be assessed for other causes of thrombocytopenia (eg, myelodysplastic syndrome, congenital thrombocytopenia, or Upshaw-Shulman syndrome).
Although dexamethasone may have prompted a more rapid response, there was concern in this older man about side effects. One alternative for achieving a rapid response is IVIG. A rapidly acting second-line therapy such as a TPO-RA or fostamatinib would be preferable to rituximab. Because of the desire for a rapid elevation in platelet count and a higher durable response rate, a TPO-RA was preferred over fostamatinib. After considering the dietary restrictions of eltrombopag, the patient preferred a once weekly romiplostim injection because his insurance did not cover avatrombopag. At a starting dose of 3 µg/kg, the platelet count increased to 14 × 103/µL after 7 days and to 26 × 103/µL by the end of the second week when the dose was increased to 5 µg/kg. According to prescribing information, it is recommended that romiplostim be initiated at 1 µg/kg per week and that the dose be escalated by 1 µg/kg per week. However, as supported by clinical trials data, we usually start at 3 µg/kg (and even higher initial doses when there is bleeding) and increase by 2 µg/kg per week.21 At week 4, the platelet count reached 64 × 103/µL, and apixaban was reinitiated. Avatrombopag would have been a good alternative, as it is an oral preparation, does not interact with multivalent cations, and leads to rapid elevation of platelet count. At day 8, a platelet response has been seen in 65% of avatrombopag-treated patients.31
Patient 4: patient needing elective surgery
The risk of bleeding in such a patient (relatively young, healthy, and asymptomatic), with platelet counts of >20 × 103/µL to 30 × 103/µL, is very low, and a watch-and-wait approach is consistent with all current clinical guidelines. It is often helpful to educate such a patient at the microscope and show him that, although his platelet number is low, his platelets are large and well granulated, both indicators of good platelet function. However, even large platelets may not fully protect him from bleeding with surgery. There are no data to predict the bleeding risk for any procedure in a patient with thrombocytopenia. The IC report suggested a target platelet count >100 × 103/µL for a neurosurgical procedure, such as the planned laminectomy in our patient.8 In emergency procedures, IVIG may rapidly increase the platelet count, but platelet transfusions are unreliable. However, our patient was scheduled for elective surgery. Corticosteroids would certainly work but had previously precipitated diabetes, and many surgeons fear that it impairs wound healing and may promote postoperative infection. IVIG is always an option but is expensive and may require multiple infusions. A recent RCT found that preoperative oral eltrombopag was not inferior to IVIG in raising the platelet count before surgery.59 In our patient we chose romiplostim 3 µg/kg each week for 2 weeks, based on a retrospective study that showed that this dose increased the platelet count to >100 × 103/µL in 79% of patients within 14 days.60 Our patient’s platelet count rose to 175 × 103/µL by the time of his operation, 17 days after initiation of romiplostim. His surgery was uneventful, and 3 weeks later, his platelet count had returned to 37 × 103/µL.
Patient 5: patient seeking a cure
Although current clinical guidelines eschew using corticosteroids for 9 months, as this patient had experienced, he was one of ∼30% of adults who had a corticosteroid remission lasting for 3 years. No clear triggering event precipitated his relapse. He was opposed to further corticosteroids, given his previous poor quality of life during the treatment. Although he had no bleeding symptoms, it was felt that he could not function at his job as an airline pilot. He was told that splenectomy had a remission rate of ∼70% but according to current clinical guidelines, he was encouraged to consider medical therapy first with either a TPO-RA or rituximab, which had durable response rates of ∼65% and ∼40%, respectively,9 but treatment-free remission rates with either is substantially lower than with splenectomy. Because of his occupation as a pilot with irregular working hours, the patient was not enthusiastic about a TPO-RA and sought a treatment that would provide the highest immediate rate of successful remission. He opted for splenectomy because of his desire not to take medications for a prolonged time, the high remission rate, and possibly the reduced need for platelet count monitoring and doctor visits. Splenectomy complications of infection and thrombosis were reviewed with him. He received pneumococcal, meningococcal, and Haemophilus influenzae type-B vaccines, and laparoscopic splenectomy was scheduled after 6 weeks. In the meantime, romiplostim was initiated as a bridging therapy before splenectomy to provide a preoperative platelet count of >50 × 103/µL. His platelet count increased to 72 × 103/µL on the day of splenectomy and was 223 × 103/µL 6 days later when romiplostim was discontinued. Thromboprophylaxis was given for 2 weeks after surgery, recognizing that some reports have shown a postsplenectomy portal and splenic vein thrombosis rate of up to 74%.61 Unfortunately, over the next 4 months, his platelet count returned to 12 × 103/µL, and he was treated with avatrombopag with a good response.
His disease course brings to light several questions.
Approximately 20% to 30% of patients fail to respond to splenectomy. Unfortunately, there is no reliable, readily available, noninvasive means of predicting the response to splenectomy. Patients with younger age (<50-60 years) and a limited number of previous therapies (≤1) have higher response rates.26,62 Platelet sequestration studies are complicated, and limited data are available, but they may aid in selecting patients who may have a favorable long-term response to splenectomy. Patients with predominantly splenic sequestration have a fivefold to sevenfold higher probability of responding to splenectomy compared with those with purely or mixed hepatic sequestration.63
The high response and remission rates for splenectomy are based on old studies when splenectomy was often performed in the first few months of disease, thereby including up to 30% of patients who would have had a remission in the first year. Therefore, response rates in patients in whom second-line medical therapies have already failed may be <70% to 80%.
It is uncertain what platelet count is adequate for laparoscopic splenectomy. Most clinical guidelines encourage efforts to increase the platelet count to ≥50 × 103/µL if possible, although successful laparoscopic splenectomy can be performed by experienced surgeons at lower counts.
Conclusion
Several new and effective ITP therapies have been introduced during the past decade, and our understanding of the disease burden and its effect on quality of life has expanded. Both of these have significantly influenced our current approach to management of ITP. Corticosteroids, at one time given for many months, are now strongly recommended to be discontinued by 6 to 8 weeks. Splenectomy, the standard second-line therapy for decades, is now recommended to be delayed for at least 1 to 2 years, allowing for more patients to achieve remission with medical therapies before considering a surgical option. As newer medications are continuously being developed and evaluated in randomized clinical trials, it is highly recommended that therapies with RCT evidence be tried first, leaving therapies with less validation for later use. Unfortunately, there are still no reliable biomarkers that can be used to guide treatment. However, current therapeutic options have variable efficacy, safety profiles, mechanisms of action, and modes of administration. As a result, an individualized approach to treatment is advised, where patient involvement, preferences, and values become central to the choice of therapy.
Authorship
Contribution: All authors contributed to the design of the study, wrote the manuscript, edited it, and agreed to its submission.
Conflict-of-interest disclosure: W.G. has received research grants from Bayer and Pfizer and consulting fees and lecture honorariums from Novartis, Amgen, Principia, Sanofi, Pfizer, Bayer, MSD and Sobi. T.G. has received research funding from the National Heart, Lung, and Blood Institute, Rigel, and Principia and consulting fees from Dova, Amgen, Rigel, Novartis, Sanofi, Cellphire, the Platelet Disorder Support Association, Bioverativ, Momenta, and Vertex. D.J.K. has received research funding from Actelion (Syntimmune), Agios, Alnylam, Amgen, Argenx, Bristol Myers Squibb (BMS), Immunovant, Kezar, Principia, Protalex, Rigel, and Takeda (Bioverativ); consulting fees from Actelion (Syntimmune), Agios, Alnylam, Amgen, Argenx, Bristol Myers Squibb (BMS), Caremark, CRICO, Daiichi Sankyo, Dova, Genzyme, Immunovant, Incyte, Kyowa-Kirin, Merck Sharp Dohme, Momenta, Novartis, Pfizer, the Platelet Disorder Support Association, Principia, Protalex, Protalix, Rigel, Sanofi, Genzyme, Shionogi, Shire, Takeda (Bioverativ), UCB, Up-To-Date, and Zafgen.
Correspondence: Waleed Ghanima, Department of Research, Østfold Hospital, PB-300 1714 Grålum, Norway; e-mail: waleed.ghanima@so-hf.no.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal