


Abstract
Pure red cell aplasia (PRCA) is a rare hematological disorder with multiple etiologies. The multifaceted nature of this disease is emphasized by the variety of concomitant clinical features. Classic idiopathic presentation aside, prompt recognition of pathogenetic clues is important because of their diagnostic and therapeutic implications. As a consequence, treatment of PRCA is diverse and strictly dependent on the presented clinical scenario. Here, we propose a series of clinical vignettes that showcase instructive representative situations derived from our routine clinical practice. Using these illustrative clinical cases, we review the diagnostic workup needed for a precise diagnosis and the currently available therapeutic options, discussing their applications in regard to the various PRCA-associated conditions and individual patients’ characteristics. Finally, we propose a treatment algorithm that may offer guidance for personalized therapeutic recommendations.
Introduction
Pure red cell aplasia (PRCA) is a rare bone marrow (BM) disorder characterized by malproductive, reticulocytopenic, normocytic anemia and the disappearance of erythroid precursors.1 Nosologically, among hematological diseases, PRCA resides within single-lineage cytopenias, sharing clinical features with BM failure syndromes and autoimmune cytopenias.2 Although the term PRCA mostly refers to acquired disease entities, Diamond-Blackfan anemia (DBA) is a rare inherited form of PRCA resulting from autosomal dominant mutations in ribosome biogenesis genes.3 Its manifestation may be occasionally delayed, but the majority of patients with DBA present in infancy or early childhood with variable severity.4 A special infectious-acquired form of PRCA is B19 parvovirus-mediated transient aplastic crisis (TAC), seen in the context of immunodeficiency (eg, AIDS, severe combined immunodeficiency), or in patients with hereditary hemolytic anemias with erythroid hyperplasia of the marrow.5,6 It is likely that some cases of PRCA, especially in the context of immunodeficiency, may also have causes besides B19-related viral etiology, but aside from vague implications of Epstein-Barr virus and cytomegalovirus (CMV) infections, the specific agents remain elusive.
DBA and virally mediated TAC aside, most of the acquired PRCA cases, particularly in adults, are idiopathic and believed to be a T cell–mediated process resulting from unknown triggers.7 Indeed, causative antibodies indicative of an autoimmune process and potential pathogenic culprits would have to recognize antigens restricted only to early erythroid precursors and absent from mature red cells to explain the lack of a hemolytic component. A particular and very rare case of acquired PRCA is linked to the use of recombinant human erythropoietin (rhEPO), reported in the early 1990s in patients treated with this drug.8-10 Subsequent epidemiologic studies revealed that this phenomenon was linked to the use of leachates in uncoated rubber stoppers of the prefilled syringes.7 Once this was recognized and resolved, rhEPO-associated PRCA became exceedingly rare.11 Besides rhEPO, several other drugs (eg, diphenylhydantoin, rifampicin) have been anecdotally reported as triggers of immunoglobulin G (IgG)-mediated, hapten-based, or directly toxic mechanisms of erythropoiesis inhibition.7,12,13 Finally, PRCA can be associated with various other diseases, including T-cell large granular lymphocyte leukemia (LGL), thymoma, and Good syndrome (ie, hypoglobulinemia and thymoma).7,14,15 The classic dyad of PRCA and thymoma (in some cases, a triad including myasthenia gravis) also has clinical implications as to the management of such patients (Figure 1).16

Common PRCA-associated disorders. Idealized circle plot showing PRCA and associated diseases according to our internal cohort of patients with PRCA, LGL, thymoma, and Good syndrome, and bar graph demonstrating frequencies of associations in our internal cohort of patients with PRCA. PB19, parvovirus B19.
Common PRCA-associated disorders. Idealized circle plot showing PRCA and associated diseases according to our internal cohort of patients with PRCA, LGL, thymoma, and Good syndrome, and bar graph demonstrating frequencies of associations in our internal cohort of patients with PRCA. PB19, parvovirus B19.
Pathogenesis of acquired PRCA: clues to effective therapies
Understanding the pathogenesis of PRCA is important for the development of treatment strategies. For instance, responses to a specific treatment modality point toward different pathogenic mechanisms. Indeed, sensitivity to T cell–targeted immunosuppression in idiopathic PRCA indicates a self-reactive T cell–mediated mechanism, whereas the invariant success of immunoglobulin supplementation (IVIG) in TAC confirms the diagnosis. Consequently, occasional responsiveness of TAC to IVIG in cases negative for B19 parvovirus may indicate the pathogenetic role of not yet recognized viral infections.17 In fact, at least theoretically viral infections may affect erythroid precursors also in an antibody-mediated fashion. Evidence derived from marrow culture studies, used in the past as a screening parameter for undiagnosed cytopenias, showed that both antibodies and T lymphocytes may mediate the inhibition of erythropoiesis in patients with PRCA and that a higher burst forming unit-erythroid number in vitro is associated to a better responsiveness to immunosuppressive treatment.18-21
In general, majority of acquired PRCA cases are idiopathic and likely due to cytotoxic T cell–mediated destruction of early erythroid precursors.1,7,22 Certain clinical associations may be very illustrative of the general pathophysiologic mechanism operating in otherwise idiopathic cases. For instance, T-LGL, a clonal lymphoproliferation of cytotoxic T lymphocytes (CTLs), is observed in 15% to 20% of patients with PRCA.23 It has been hypothesized that such a clonal process is the result of a highly specific recognition of an immunodominant antigenic peptide on erythroid precursors originating from initially physiologic polyclonal reactions.20,21 The fidelity of the lineage restriction of this process implies that the main effector mechanism of cytotoxicity is mediated by the T-cell receptor (TCR) rather than soluble cytotoxic cytokines (eg, interferon-γ, Fas ligand; both of which would be expected to cause a less lineage-selective damage).23 T-LGL association may represent an extreme pole of the continuum pathologic spectrum of CTL responses: from polyclonal in classic PRCA to oligoclonal and then clonal in the LGL-associated variant.24 However, as in many autoimmune diseases, including aplastic anemia (AA), the antigens remain elusive. The corresponding inciting events may include molecular mimicry leading to a breach of tolerance with immune responses resulting as a cross-reactive TCR recognition (Figure 2). A minority of clonal CTL expansions is also associated with proliferation of γ/δ T cells.25-27 These γ/δ T lymphocytes lack HLA class I restriction and use killer cell inhibitory receptors to exert cytotoxic activities.28 Cases of PRCA associated with γ/δ LGLs have been described and share similar clinical features with typical α/β LGL proliferation.25,26,29 The targeting of erythroid progenitors in γ/δ LGL cases could reflect the physiologic downregulation of HLA class I antigens in the erythroid progenitor cells.29
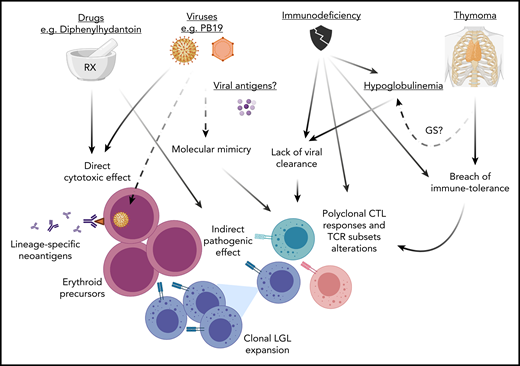
Pathogenesis of PRCA. Various mechanisms may lead to PRCA. (1) Viral infections (eg, B19 parvovirus) and a variety of drugs may have a direct cytotoxic effect against erythroid precursors or also exert an indirect pathogenic effect via a molecular mimicry mechanism. The resulting cross-reactive TCR recognition may lead to a breach of tolerance with polyclonal CTL responses. (2) This latter phenomenon may also be triggered by alteration of immune tolerance because of the presence of thymoma (or thymic dysfunction) with subsequent LGL clonal expansion (in some cases carrying STAT3 mutations). (3) Lack of viral clearance from immunodeficiency (eg, hypoglobulinemia) may be the cause of elicitation of aberrant CTLs responses. Alternatively, in the context of immunodeficiency antibody responses or the inability of TCR recognition may lead to generation of autoreactive T cells with alterations of TCR repertoires evading immune-tolerance mechanisms. For instance, the presence of T-cell subsets with predominant expression of γδ+ TCR rearrangements lacking HLA class I restriction could explain the attack of erythroid progenitors, which physiologically present downregulation of HLA class I antigens. GS, Good syndrome; STAT3, signal transducer and activator of transcription 3.
Pathogenesis of PRCA. Various mechanisms may lead to PRCA. (1) Viral infections (eg, B19 parvovirus) and a variety of drugs may have a direct cytotoxic effect against erythroid precursors or also exert an indirect pathogenic effect via a molecular mimicry mechanism. The resulting cross-reactive TCR recognition may lead to a breach of tolerance with polyclonal CTL responses. (2) This latter phenomenon may also be triggered by alteration of immune tolerance because of the presence of thymoma (or thymic dysfunction) with subsequent LGL clonal expansion (in some cases carrying STAT3 mutations). (3) Lack of viral clearance from immunodeficiency (eg, hypoglobulinemia) may be the cause of elicitation of aberrant CTLs responses. Alternatively, in the context of immunodeficiency antibody responses or the inability of TCR recognition may lead to generation of autoreactive T cells with alterations of TCR repertoires evading immune-tolerance mechanisms. For instance, the presence of T-cell subsets with predominant expression of γδ+ TCR rearrangements lacking HLA class I restriction could explain the attack of erythroid progenitors, which physiologically present downregulation of HLA class I antigens. GS, Good syndrome; STAT3, signal transducer and activator of transcription 3.
Another clue as to the pathogenesis of PRCA stems from the association of PRCA with hypoglobulinemia with thymoma (Good syndrome) or without thymoma (common variable immunodeficiency [CVID]). The latter phenomenon implies that antibody responses or the inability of TCR recognition may lead to overshooting CTL responses with cross-reactivity directed to red cell precursors.21,30 Alternatively, deficient humoral responses in patients with hypoglobulinemia may lead to viral persistence or inability to clear infectious agents with either direct toxicity or induction of cross-reactive polyclonal T-cell responses.31 It is possible that cases of PRCA seen in the context of B-cell lymphoproliferative disorders may indeed be due to either disease-intrinsic or iatrogenic hypoglobulinemia. The occasional responsiveness to IVIG is not completely understood and implies either clearance of a cryptic viral trigger or masking of the autoimmune epitopes as proposed in immune thrombocytopenic purpura.32 The pathogenetic mechanisms behind the association of thymoma and PRCA remain elusive, and a faulty elimination of autoimmune clones or the alteration of other mechanisms of immune tolerance could be stipulated.33
A specific remark has to be made for the case of PRCA arising in the context of allogeneic ABO-incompatible stem cell transplantation (allo-SCT). This scenario is observed in case of major ABO incompatibility identified most commonly by the combination of a blood group O recipient and a blood group A, B, or AB donor, occurring at a variable frequency (7% to 29%) according to the type of conditioning regimen used.34,35 The persistence of host antidonor iso-hemagglutinins is responsible for delayed erythroid recovery.36 This type of PRCA is often self-limiting and managed with transfusion support but in some cases may persist for more than 6 months, requiring specific treatments.37
Case 1: initial workup and first-line treatment of idiopathic PRCA
A 33-year-old African American woman was admitted after presenting with recent onset of shortness of breath and a hemoglobin (Hb) level of 4.8 g/dL with no other complete blood count (CBC) abnormalities. Her medical history was unremarkable, apart from flu-like symptoms and generalized fatigue dating 2 weeks before her hospital admission. On physical examination, the patient was afebrile, showed mild tachycardia (106 beats/min) and no other physical findings. An initial laboratory workup showed extremely low reticulocytes count (<10 000/µL), normal iron indices and negative Coombs test and renal and hepatic function. The patient was administered red cell transfusions and her condition soon stabilized.
Diagnostic procedures of a suspected PRCA case
Congenital BM failure aside, profound reticulocytopenia (<10 000/µL) and exclusion of other causes of malproductive normocytic, normochromic anemia (metabolic deficiencies, systemic diseases) in the context of an otherwise normal CBC are a prerequisite for PRCA7,22 (Table 1). The presence of other cytopenias, unless otherwise explained by the presence of concomitant or associated conditions, is incompatible with the diagnosis of PRCA, which is established on morphologic BM finding of erythroid hypoplasia (<1% to 5% of total cellularity and increased myeloid:erythroid ratio) with normal morphology and maturation of other lineages.22 Usually, the arrest of erythroid differentiation is complete and all erythroid progenitor cells are absent. However, erythroid hypoplasia may also be occasionally found in some cases of myelodysplastic syndrome (MDS), but the residual erythroblasts are usually dysplastic as well as the other lineages.17 Nutritional deficiencies have to be excluded. Indeed, erythroid hypoplasia may be encountered in copper deficiency or rare presentations of vitamin deficits.22 Certain morphologic features, such as myeloid and erythroid precursors vacuolization and occasional ring sideroblasts, may provide clues as to specific etiology (eg, in case of hypocupremia). Of note is that copper deficiency may be associated with neutropenia and dyserythropoiesis with a typically normal platelet count.38 In PRCA, the BM is mostly normocellular and cytogenetics normal. Somatic mutational panels are usually negative for molecular lesions typical of myeloid neoplasia, but mutation frequency may be as high as 53% according to the number and type of genes tested (especially if including BM failure genes).39,40 In particular, STAT3 mutations (eg, in order of frequency Y640F, D661Y, D661V, N647I), typical of LGL, may be found in cases associated with this condition.41-43 Indeed, coincidental findings may include polyclonal T-cell infiltrate or the presence of an actual LGL diagnosis. Erythropoietin levels are usually high or very high.44 Active viral infections should be ruled out. Moreover, patients may have hypoglobulinemia or coincidental monoclonal gammopathy of undetermined significance or chronic lymphocytic leukemia, which do not preclude a PRCA diagnosis.45
Differential diagnoses of reticulocytopenic anemias
| Category | Conditions | Examples/associations |
|---|---|---|
| Intrinsic bone marrow disorders | Acquired PRCA | LGL, thymoma, CVID, PB19, lymphoproliferative disorders, ABO-incompatible allo-SCT |
| Congenital PRCA | Diamond-Blackfan anemia | |
| Acquired AA | Idiopathic, postviral | |
| Congenital AA | Fanconi anemia | |
| Systemic | Acute/chronic infections | HIV, EBV |
| Nutritional | Iron, folic acid, vitamin B12 | |
| Metabolic | Hypothyroidism, chronic kidney failure | |
| Autoimmune disorders | Ulcerative colitis, Crohn disease, RA | |
| Drugs | Linezolid, chemotherapy | |
| Neoplastic | Hematologic malignancies | AML, MDS, myelofibrosis (spent phase of MPN) |
| Metastatic solid cancers | Prostate, breast cancer |
| Category | Conditions | Examples/associations |
|---|---|---|
| Intrinsic bone marrow disorders | Acquired PRCA | LGL, thymoma, CVID, PB19, lymphoproliferative disorders, ABO-incompatible allo-SCT |
| Congenital PRCA | Diamond-Blackfan anemia | |
| Acquired AA | Idiopathic, postviral | |
| Congenital AA | Fanconi anemia | |
| Systemic | Acute/chronic infections | HIV, EBV |
| Nutritional | Iron, folic acid, vitamin B12 | |
| Metabolic | Hypothyroidism, chronic kidney failure | |
| Autoimmune disorders | Ulcerative colitis, Crohn disease, RA | |
| Drugs | Linezolid, chemotherapy | |
| Neoplastic | Hematologic malignancies | AML, MDS, myelofibrosis (spent phase of MPN) |
| Metastatic solid cancers | Prostate, breast cancer |
AML, acute myeloid leukemia; EBV, Epstein-Barr virus; MPN, myeloproliferative neoplasm; PB19, B19 parvovirus; RA, rheumatoid arthritis.
All these additional findings may be of importance for selection of proper therapies. In case of TAC, the presence of giant pronormoblasts and parvovirus B19 polymerase chain reaction positivity confirms the diagnosis.5,46 Serology may also be helpful: usually, B19 parvovirus IgG presence excludes the diagnosis of TAC because humoral immunity is protective, whereas positivity for IgM alone may indicate early infection.47 However, in a proper setting, testing for immunodeficiency should be performed (HIV, CVID) because, if severe, it may render serologic diagnosis uninformative.47 Moreover, testing for hemolytic anemia resulting from hemoglobinopathy or autoantibodies should be performed when appropriate. Exclusive conditions include renal failure, myeloid neoplasia, and AA. Thus, once the diagnosis of PRCA is made, the workup should include testing for the presence of LGL (flow cytometry, TCR rearrangement, morphologic enumeration of LGL), chest computed tomography (CT) scan to rule out thymoma, immunoglobulin levels, and, if appropriate, testing for hemoglobinopathies or congenital hemolytic anemias (eg, spherocytosis) (Table 2). Finally, even if rare in adulthood, the possibility of a DBA diagnosis should be excluded. In particular in cases with young age at presentation and macrocytosis, elevated erythrocyte adenosine deaminase levels registered before red cells transfusions may be useful to identify such patients.48 Further genetic testing for “DBA genes” confirms the diagnosis.49,50
List of initial workup studies for patients presenting with a possible diagnosis of pure red cell aplasia
| Studies |
|---|
| Anemia |
| CBC with differential |
| Reticulocyte count |
| EPO levels |
| Coombs test |
| Nutritional deficiencies |
| Hb electrophoresis and peripheral blood smear examination |
| Diamond-Blackfan anemia |
| Erythrocyte ADA (before transfusion of red cells) |
| Genetic testing |
| LGL |
| Flow cytometry (as part of a low-grade lymphoma panel) |
| TCR rearrangement |
| STAT3 gene mutations (eg, in order of frequency Y640F, D661Y, D661V, N647I) |
| Autoimmunity and immunologic conditions |
| Antinuclear antibodies |
| Rheumatoid arthritis antibodies |
| Extractable nuclear antigen panel |
| Lymphocyte subsets (flow cytometry) |
| B-cell dyscrasias studies |
| Serum electrophoresis |
| Serum immunofixation |
| Free light chains |
| Immunoglobulins levels |
| Bone marrow |
| Morphology (myeloid:erythroid ratio, dysplastic changes, giant pronormoblasts) |
| Flow cytometry for LGL |
| Cytogenetics |
| TCR rearrangement |
| Virological |
| B19 parvovirus |
| Viral hepatitis |
| HIV |
| EBV |
| CMV |
| Anamnestic data about drug history |
| Thymoma studies* |
| CT scan of the chest |
| Studies |
|---|
| Anemia |
| CBC with differential |
| Reticulocyte count |
| EPO levels |
| Coombs test |
| Nutritional deficiencies |
| Hb electrophoresis and peripheral blood smear examination |
| Diamond-Blackfan anemia |
| Erythrocyte ADA (before transfusion of red cells) |
| Genetic testing |
| LGL |
| Flow cytometry (as part of a low-grade lymphoma panel) |
| TCR rearrangement |
| STAT3 gene mutations (eg, in order of frequency Y640F, D661Y, D661V, N647I) |
| Autoimmunity and immunologic conditions |
| Antinuclear antibodies |
| Rheumatoid arthritis antibodies |
| Extractable nuclear antigen panel |
| Lymphocyte subsets (flow cytometry) |
| B-cell dyscrasias studies |
| Serum electrophoresis |
| Serum immunofixation |
| Free light chains |
| Immunoglobulins levels |
| Bone marrow |
| Morphology (myeloid:erythroid ratio, dysplastic changes, giant pronormoblasts) |
| Flow cytometry for LGL |
| Cytogenetics |
| TCR rearrangement |
| Virological |
| B19 parvovirus |
| Viral hepatitis |
| HIV |
| EBV |
| CMV |
| Anamnestic data about drug history |
| Thymoma studies* |
| CT scan of the chest |
Particulary in case of history of myasthenia gravis.
ADA, adenosine deaminase; EBV: Epstein-Barr virus; EPO, erythropoietin; STAT3, signal transducer and activator of transcription 3.
Back to our case 1: evaluation and first-line treatment
BM evaluation of our patient showed normal cellularity for age (70% to 80%), paucity of erythroid precursors (<1% by manual count) with unremarkable granulocytic and megakaryocytic lineage, and no blasts nor atypical cells. Cytogenetic evaluation showed a normal female karyotype. Flow cytometry revealed no immunophenotypic evidence of a lymphoproliferative disorder and immunoglobulin levels were normal. Chest CT scan ruled out the presence of thymoma, and virologic/serologic studies were all negative. Therefore, a diagnosis of idiopathic acquired PRCA was made.
Considerations and treatment strategy
Despite being highly effective, high-dose corticosteroids do not produce durable responses and have important long-term side effects.1 The most effective first-line treatment of idiopathic PRCA is cyclosporine A (CsA) administered at a starting dose of 2 to 6 mg/kg per day (in divided doses) possibly combined with steroid (prednisone at 30 mg/day) with a rapid taper, yielding an overall response rate (ORR) of about 65% to 87%.14,17,51-54 Because of the known nephrotoxic potential of calcineurin inhibitors, periodic monitoring of renal function is recommended. Moreover, taking into consideration inter-individual variability of cytochrome P450 isoenzymes and drugs interactions, CsA concentrations should be monitored and dose adjusted to the therapeutic levels of 150 to 250 ng/mL.7 However, responses can be seen also without achieving these concentrations and can be faster with corticosteroids, becoming stable usually not earlier than 6 weeks and up to 3 months from treatment start.55 Once a sustained Hb normalization is attained (1-2 months), we prefer to continue the treatment for 1 to 2 additional months. A slow CsA taper can be then initiated in an attempt to establish the minimal effective dose (maintenance) able to prevent relapses.56 Maintenance should include CsA, albeit its combination with steroids demonstrated better relapse-free survival (103 vs 33 months, P < .01).55 In general, relapses are common, and careful tapering of CsA allows for early detection. Patients who stop the treatment without appropriate tapering are at risk for relapse occurring at a median time of 3 months (1.5-40) from drug withdrawal.54 To limit long-term iatrogenic complications, a judicious follow-up is required and a minimal maintenance dose should be established.57 Apart from CsA, tacrolimus has also been used in patients who fail to respond or did not tolerate CsA, but its use upfront has been limited because of anecdotal evidence of triggering PRCA.58,59 Of note is that the selection of first-line treatment has to consider comorbidities (eg, chronic kidney disease) or other precluding factors.
Case 2: PRCA associated with LGL in the elderly
An 80-year-old man was referred to his cardiologist because of progressive dyspnea. He had a history of coronary artery disease treated with bypass surgery 10 years before and chronic kidney disease stage III. CBC showed severe anemia (Hb 3.4 g/dL, hematocrit 16.2%, mean corpuscular volume 90.3 fL) and reticulocytopenia, a relative lymphocytosis (51%) with absolute lymphocyte count of 2.8 × 109/L and a mild neutropenia (1.02 × 109/L). Creatinine level was 1.6 mg/dL with normal hepatic function. The patient received blood transfusions and was subsequently referred to our attention for a hematology consult. A BM biopsy demonstrated increased population of CTLs representing ∼60% of BM cellularity with very few erythroid progenitors. Cytogenetic and fluorescence in situ hybridization studies for MDS were normal. Flow cytometry revealed an abnormal population of T cells expressing CD3, CD8, and CD57. Although STAT3 mutational analysis was negative, TCR gene rearrangement showed a clonal pattern. These findings were consistent with a concomitant diagnosis of T-LGL.43 Taking into consideration his renal function and the diagnosis of LGL-associated PRCA, our patient was started on oral cyclophosphamide (CY) 25 mg and prednisone 60 mg daily. CY was subsequently increased to 50 mg without any sign of myelotoxicity, prednisone was tapered off over a month, and response was soon heralded by reappearance of reticulocytes.
Considerations and treatment strategy
The immunomodulatory properties of cytotoxic agents (CY, methotrexate) usually in combination with steroids have been used for the treatment of PRCA, especially in refractory cases or in those with contraindications for CsA.1,17 The initial dosage of CY is usually 50 to 100 mg per day orally with prednisone at a dose of 20 to 30 mg/day, carefully monitoring CBC to avoid myelotoxicity.7 ORR of 40% to 60% has been reported at a median time of 12 weeks, followed by a tapering phase, for a total time of CY treatment not exceeding 6 months.22 Maintenance therapy may prolong relapse-free survival but experiences with patients affected by Wegener’s granulomatosis, chronically exposed to CY, demonstrated an increased risk of bladder or hematological cancers and gonadal toxicity proportional to cumulative dose and duration of therapy.60,61 Thus, CY may be used under these considerations in older individuals and for a limited time in younger patients. Combination of CY plus steroids seems to be particularly effective in the context of LGL-associated PRCA cases, as shown by the prolonged remissions when compared with other treatment modalities (steroids alone, CsA, or methotrexate).14 ORR in this setting is as high as 66% to 100% with median duration of response of 32 to 53 months.62,63 Relapse seems to be less common in LGL-associated PRCA treated with CY, but the excellent median duration of response may preclude a real estimation of such complication. Moreover, relapsing patients appear to be responsive to repeated round of immunosuppression.54
Methotrexate has also been used for treating patients with PRCA associated with LGL, but response rates have been inferior to those of other treatment strategies (eg, CY, CsA). In addition, because of the interference with folate metabolism and the inherent complication of anemia, its utility has been linked mostly to the treatment of T-LGL with neutropenia.17
Case 3: thymoma-associated PRCA
A 39-year-old woman presented with generalized weakness and fatigue. Her medical history and physical examination were unremarkable. A CBC showed an Hb level of 6.7 g/dL, and reticulocyte count was markedly reduced (<10 000/µL). No other metabolic alteration or viral positivity were found at the initial laboratory workup. A BM evaluation showed normocellular marrow with a paucity of erythroid precursors, whereas a chest CT scan revealed the presence of a mediastinal mass suggestive for thymoma. The patient was then referred to the thoracic surgery department for thymoma resection.
Considerations and treatment strategy
After LGL, thymoma is the second most frequent disorder associated to PRCA.7 Among patients with thymoma, PRCA seems to be present in around 5% to 7% of cases.33 When considering patients with PRCA, the frequency of thymoma coincidental diagnosis is as high as 30%, as recently shown by a review of our internal cohort.15 In these cases, thymectomy has been invoked as an initial curative treatment with an expected response in up to one-third of patients.1,64 However, these results have not been confirmed in more recent reports, showing lower response rates without additional immunosuppressive treatment.52,65 Moreover, the evidence of development of PRCA after thymoma removal (at a median time of 80 months) presents another possible scenario and provides clues as to the pathogenetic mechanism underlying this condition.16 Thymic dysfunction inherent to thymoma may possibly be responsible for selection of TCR repertoires and the inability to delete autoaggressive CTLs, or for lack of effective immune responses and subsequent poor clearance of viral antigens.66-68 This latter hypothesis is also supported by our recent findings regarding the overlap of PRCA and LGL in patients affected by Good syndrome.15 Hypoglobulinemia alone or in combination with B- or T-cell lymphoproliferative disorders may play a role in causing an inability to eliminate inciting antigens and overcome infections, thereby creating a propensity to overshooting compensatory CTL responses. Besides surgery, therapeutic approaches of thymoma-associated PRCA include CsA-containing regimens, which provide high efficacy in terms of response rate and relapse-free survival.16,65
Case 4: refractory PRCA
A frail 75-year-old man with CHF (New York Heart Association class II; left ventricular ejection fraction 40%69 ) and a positive stress test presented with a history of refractory PRCA dating 3 years before, and a concomitant STAT3Y640Fmutant T-LGL diagnosis. Initial therapy included corticosteroids, followed by CsA and CY, which did not result in a response and/or were not tolerated. The patient was considered a poor candidate for anti-thymocyte globulin (ATG). Following testing for CMV and Epstein-Barr virus DNA to exclude persistent viremia, a decision was made to initiate therapy with alemtuzumab at a dose of 10 mg subcutaneously once weekly, together with adequate anti-infective prophylaxis (trimethoprim-sulfamethoxazole, acyclovir) and CMV monitoring.
Considerations and treatment strategy
Therapy of refractory PRCA may constitute a conundrum for the managing hematologist. In the presented case, the patient has a diagnosis of LGL-associated PRCA with STAT3Y640F mutation. Somatic mutations of this gene have been identified in 40% of patients with LGL and also in patients with PRCA irrespective of a concomitant LGL diagnosis.42,43 In particular, STAT3 mutations have been linked to a young age of PRCA onset and refractoriness to CsA, as in our presented case.42
Alemtuzumab has been used in the therapy of both refractory LGL and PRCA cases to provide a more profound immunosuppression.70-74 However, early applications of this drug at doses used for B-cell lymphomas (90 mg intravenously per week) led to significant toxicity and infectious complications.72 Following this schedule, alemtuzumab has also been used in intravenous regimens for the therapy of LGL (at a 10 mg daily infusion for 10 days doses with the option of a second cycle).75 A good efficacy and tolerability of a subcutaneous dosing regimen has been demonstrated in AA and single-lineage marrow failure.76,77 A first pilot study showed the safety and efficacy of this drug given at a dose of 73 mg subcutaneously over 4 consecutive days followed by CsA maintenance in 12 patients with PRCA with an ORR of 84%.76 We recently confirmed these results by applying a less intensive regimen of alemtuzumab at a fixed dose of 10 mg subcutaneously once/twice weekly for 4 to 8 consecutive weeks, with tapering and a maintenance dose of once/twice weekly on a monthly basis, demonstrating an ORR of 62% in refractory PRCA cases (13 idiopathic, 3 associated with thymoma, and 4 with LGL).17,77 We prefer this regimen because of its excellent tolerability and the possibility of escalating the dose to twice weekly. Patients given alemtuzumab should receive Pneumocystis jirovecii and anti-herpetic prophylaxis with trimethoprim-sulfamethoxazole and acyclovir and need to be carefully monitored for CMV reactivation. Last, lamivudine should be given in cases with occult hepatitis B viral infections. We also recommend checking for immunoglobulin levels and replace eventual hypoglobulinemia with intravenous immunoglobulin before initiation of alemtuzumab. Despite the rarity, a possible mechanism of resistance to alemtuzumab may be due to phenotypic switch and loss of CD52 expression.78,79
ATG is an alternative first-line salvage therapy for refractory PRCA, albeit less attractive than alemtuzumab in our case because of the presence of heart failure and poor performance status. Although literature reports are scarce, our experience shows an ORR of 30%.17,80,81 However, the advantages of alemtuzumab vs ATG are obvious and include cost, ease of administration, outpatient schedule, and overall tolerability.76
Conceptually, refractoriness to immunosuppressive treatment may be the result of insufficient intensity or the establishment of escape mechanisms (eg, CD52 phenotypic switch for alemtuzumab). Based on the positive results obtained in other immune-mediated cytopenias, such as autoimmune lymphoproliferative syndrome, immune thrombocytopenic purpura, and Fisher-Evans syndrome, sirolimus has been used in refractory PRCA cases with ORR of 76%.82 A more recent study also demonstrated its high efficacy (ORR 91%) in treatment-naive patients with renal insufficiency, as an alternative to CsA.83
In refractory cases with hypoglobulinemia, IVIG administration may be useful, possibly clearing hypothetical unrecognized viral antigens triggering exuberant CTL responses (eg, in autoantibody-mediated or Good syndrome cases).15,84 Moreover, younger patients with suspicion of a congenital disease may be considered eligible for danazol treatment.17 Also, translating positive experiences with primary autoimmune hemolytic anemia and thrombocytopenia, several reports used rituximab in the setting of PRCA associated with B-cell lymphoproliferative disorders (ie, chronic lymphocytic leukemia), with conflicting results.17,85,86 Last, bortezomib has been used in refractory PRCA cases with B-cell dyscrasias (monoclonal gammopathy of undetermined significance), whereas tofacitinib showed efficacy in cases with concomitant LGL and rheumatoid arthritis.17
A special consideration has to be made for thymoma-associated cases, in which relapse or refractoriness of PRCA should be considered a “red flag” for recurrence/residual thymoma prompting appropriate imaging studies.
Final considerations
PRCA is not a monolithic entity and thus its management may be influenced by the clinical context often pointing toward distinct pathogenetic mechanisms (Figure 3). In most cases, immunosuppressive treatment leads to transfusion independence. Long-term follow-up data of the Japanese PRCA Collaborative Study Group showed no differences in overall survival (OS) among the major subtypes of PRCA (idiopathic, thymoma-associated, and LGL-associated).54 However, this analysis emphasized that patients with PRCA have in general a shorter life expectancy if compared with age- and sex-matched healthy controls.54 Although not corroborated in other cohorts, this is particularly true for relapsing patients undergoing multiple rounds of immunosuppression because they are more prone to develop serious infectious complications, as well as organ failures related to transfusional iron overload. In addition to immunosuppressive drugs, allo-SCT has been described as an alternative treatment option for such relapsing cases. A recent survey of the European Group for Blood and Marrow Transplantation registry showed that among 33 adult patients with acquired PRCA and a median number of 3 (1-7) treatment lines, 5-year OS and event-free survival were 51% and 40%, respectively.87 These results are disappointing given the median age of transplanted patients (37 years), the nonmalignant nature of this condition and the results of conservative therapies. Despite this, it is noteworthy that in a subanalysis of the same study on 8 patients treated with an AA transplant conditioning strategy (BM as a source of HSC and in vivo T-cell depletion with alemtuzumab or ATG and CY), the 5-year OS and event-free survival were 75% and 47%, respectively.87 In either case, we rarely resort to transplant referral in our patients with PRCA. Therefore, the decision to proceed with allo-SCT, the correct timing and type of conditioning should be a subject of careful benefit/risk considerations because they remain controversial and explored to date only in retrospective case series.87,88

Treatment algorithm of acquired PRCA. Cs, corticosteroids; CY, cyclophosphamide; M, maintenance; MGUS, monoclonal gammopathy of undetermined significance; PB19, parvovirus B19.
Treatment algorithm of acquired PRCA. Cs, corticosteroids; CY, cyclophosphamide; M, maintenance; MGUS, monoclonal gammopathy of undetermined significance; PB19, parvovirus B19.
A special remark has to be made for patients diagnosed with PRCA at young age. Because of the possible long-term transfusion requirement, iatrogenic hemochromatosis may be expected in this particular setting, especially in cases of refractory disease with a high transfusion burden.22 However, with the exception of desferrioxamine, iron chelation may possibly interfere with calcineurin inhibitors, the standard PRCA first-line treatment. Our suggestion is to promptly start young patients on PRCA treatment and, once Hb levels are stabilized, give appropriate iron chelation treatment with subcutaneous desferrioxamine (40-60 mg/kg per day over 8-12 hours per night 4 to 7 nights per week) as the preferred agent.49 Because of the overall high success rate, causative therapy is prioritized over iron overload treatment in our practice. Iron chelation should be also considered in all refractory patients.
In summary, because of the rarity of the condition and the paucity of systematic clinical trials–derived information, the management of acquired PRCA should be based on the growing body of evidence resulting from retrospective case series and consideration of clinical clues guiding an individualized therapeutic approach.
Acknowledgments
This work was supported by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship (C.G.) and by the National Institutes of Health, National Heart, Lung, and Blood Institute (grant R35HL135795) (J.P.M.).
Authorship
Contribution: C.G. and J.P.M. conceived the idea and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jaroslaw P. Maciejewski, Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, 9620 Carnegie Ave, Building NE6-314, Cleveland, OH, 44106; e-mail: maciejj@ccf.org.
