

T-cell/histiocyte-rich large B-cell lymphoma (TCRLBCL) has a unique tumor microenvironment (TME) with an extensive infiltrate of T cells and histiocytes. In this issue of Blood, found that the unique immunoarchitecture of this TME could be an important contributor to immune escape of the lymphoma cells.1
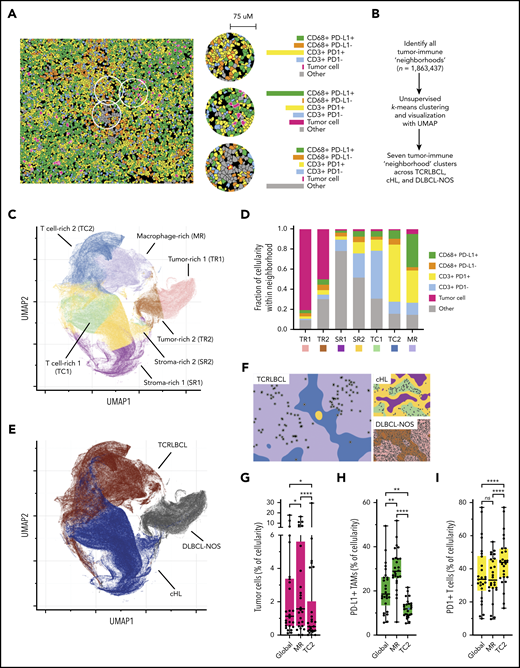
Spatially defined immune cell “neighborhoods” distinguish TCRLBCL from cHL and DLBCL-NOS. The cellular composition together with the expression of PD-L1 and PD-1 is determined in small regions to construct tumor-immune “neighborhoods” throughout the section. These neighborhoods are then clustered into 7 clusters using K-mean clustering, which are used to visualize and compare the immunoarchitecture of different cases and different types of lymphoma. See Figure 4 in the article by Griffin et al that begins on page 1353.
Spatially defined immune cell “neighborhoods” distinguish TCRLBCL from cHL and DLBCL-NOS. The cellular composition together with the expression of PD-L1 and PD-1 is determined in small regions to construct tumor-immune “neighborhoods” throughout the section. These neighborhoods are then clustered into 7 clusters using K-mean clustering, which are used to visualize and compare the immunoarchitecture of different cases and different types of lymphoma. See Figure 4 in the article by Griffin et al that begins on page 1353.
Nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) is classified as a subtype of HL. There are some major differences between NLPHL and the classical subtypes of HL (cHL), notably in the preservation of the B-cell and the germinal center B-cell differentiation program. NLPHL is generally an indolent disease with good prognosis, but it may progress to 2 forms of diffuse large B-cell lymphoma (DLBCL). One of them is characterized by a TME with an extensive infiltrate of T cells and histiocytes overshadowing the scattered tumor B cells, the TCRLBCL. The other one has a predominance of large tumor cells similar to the usual DLBCL. TCRLBCL may also present as a de novo lesion without evidence of a preexisting NLPHL, and it is not entirely clear how and if these de novo TCRLBCL are related to the TCRLBCL arising from the setting of NLPHL. Recent genomic analyses indicate significant similarity of the mutation profiles among TCRLBCL,2 DLBCL derived from NLPHL3 and the ST2 subtype of DLBCL,4 suggesting that these tumors are related and may be derived from an ancestral cell resembling the neoplastic cell of NLPHL. The TME of the TCRLBCL is unique among DLBCLs. How the tumor cells escape from immune surveillance amid all these stromal T cells is intriguing and is the focus of the study by Griffin and coworkers published in this issue of Blood.
The authors found that the malignant B cells in TCRLBCL frequently harbored PDL1/PD-L2 copy gain or amplification, which was associated with increased PD-L1 expression (P = .0111). They performed multiparametric immunophenotyping studies with sophisticated spatial image analyses and found that the malignant B cells were surrounded by a high number of PD-L1–expressing macrophages and PD-1–positive T cells. The distributions of macrophages, T cells, and B cells together with their PD-L1 or PD-1 expression (spatially defined immune cell neighborhoods) were determined and found to be rather distinct compared with the “neighborhoods” in DLBCL-NOS, and cHL, suggesting a unique functional interaction between the PD-L1–expressing cells and the PD-1–positive T cells in TCRLBCL (see figure). The authors then examined data on patients who were enrolled in clinical trials of PD-L1/PD-1 blockade for refractory hematologic malignancies. They found 5 patients with relapsed/refractory TCRLBCL and observed clinical response in 3 of the 5 patients, including 2 complete responses and 1 partial response. The clinical observation, although preliminary, provided support that PD-L1/PD-1 interaction in the TME is clinically important and could be a target for therapeutic intervention.
Although an increase in the copy number of PD-L1/PD-L2 in the tumor cells is associated with an increase in expression of these genes, PD-L1 was also highly expressed in stromal macrophages, which the authors attributed to the upregulation of the JAK/STAT1 pathway in macrophages through interferon-γ stimulation. JAK2 is part of the 9q34 amplicon, and its expression has been shown to be increased with copy number gain. This may lead to enhanced JAK2/STAT signaling and JAK2-mediated nuclear chromatin remodeling.5 Mutations in the tumor may also contribute to enhance JAK/STAT pathway activation. Thus, the cytokine environment may be modified by these genetic changes, and PD-L1 expression in stromal macrophages may be regulated through a more complex cytokine network.
It is plausible that the PD-L1/PD-1 interaction is an important mechanism in immune escape as proposed in Griffin’s article. However, as in cHL and other lymphomas, response to PD-L1/PD-1 immune check point blockade may be partial or absent among patients. Thus, the biological determinants that affect therapeutic efficacy need to be further explored. These include other immune checkpoints that are not measured, other immunosuppressive cells, and immunomodulatory compounds in the TME, the abundance, composition, and responsiveness of immune effector cells, and the intrinsic characteristics of the tumor cells. With technical and analytical advances, many of these parameters and their potential interactions can now be investigated.
The multiparameter immunophenotyping approach described in Griffin’s work provided useful and informative data. There are additional exciting developments that can move the field further forward. The time of flight mass cytometry technology,6 originally developed for flow-based assays, has been adapted to study tissues in situ (imaging mass cytometry). This technology allows the assessment of far more markers than possible using fluorescence-based assays. This and other high-dimensional imaging approaches have been reviewed recently.7
Gene expression profiling studies on lymphoma have traditionally been performed on bulk tumor RNA. Recent development in computational analysis, such as the CiberSort approach,8 may help to deconvolute bulk GEP data to provide an estimate of the immune cell populations present in the TME. Single-cell (sc) RNA-seq approaches9 have been employed recently to decipher the biological complexity of the tumor cell as well as the stromal cell populations. When scRNA-seq is performed on isolated cells, spatial information is lost, and cell isolation procedures may introduce various artifacts. To overcome these barriers, techniques such as Slide-seq,10 that attempt to preserve the spatial information at single-cell resolution, are being developed. This will add a whole new dimension to immunophenotyping.
Using these innovative cutting-edge technologies, it is feasible to comprehensively analyze the tumor with its various components and their interactions. However, they have to be coupled with well-designed studies with sufficient number of well-annotated patients to answer critical clinical questions, such as the determinants of response vs resistance to immunotherapy, the prediction of the most effective single or combinations of agents for each patient, and how to manipulate the system to enhance therapeutic efficacy and overcome resistance. We could anticipate new and exciting discoveries in immunotherapy in the near future.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal