


Abstract
Treatment of relapsed or refractory acute myeloid leukemia (AML) has presented challenges for hematologists for decades. Despite numerous clinical studies, outcomes are consistently disappointing with 5-year overall survival rates of ∼10%. Allogeneic hematopoietic cell transplantation at the time of second complete remission remains the only reliable option with curative potential. However, recent approval of several new agents has transformed treatment paradigms that had been in place for almost half a century in AML. This new therapeutic landscape provides the opportunity to revisit the approach to relapsed or refractory AML. Through illustrative cases, we describe our approach, which increasingly relies on specific disease biology. We focus on treatment outside of the context of clinical trials because such trials are not available in most parts of the world. Primarily, we consider age, fitness to tolerate intensive chemotherapy, remission duration, and presence of a targetable mutation to guide treatment. The coming years will inevitably bring new targets and agents that may prove most effective when combined with each other and/or chemotherapy. Future studies are needed to determine how best to implement this evolving armamentarium of treatment options, to elucidate mechanisms of resistance, and to continue the pursuit of novel drug discovery.
Introduction
Effective treatment of relapsed or refractory acute myeloid leukemia (AML) has presented a Sisyphean challenge for hematologists for decades. Despite remarkable insights into deciphering the molecular pathogenesis of AML and important albeit modest advances in treatment, >10 000 of the ∼20 000 new patients with AML diagnosed in the United States each year die of their disease.1 Relapse is the most common cause of treatment failure. The 5-year overall survival (OS) for adult patients with AML (non–acute promyelocytic leukemia [non-APL] AML) after disease relapse is only 10% (Figure 1) in both older studies with long-term follow-up2-4 and in more contemporary series,5,6 although available data are from studies prior to the promising era of molecular genetics and targeted therapy. Furthermore, ∼20% of patients demonstrate primary induction failure (PIF; further defined in the next section).7,8 Currently, allogeneic hematopoietic cell transplantation (HCT) is the only reliable option with curative potential, with OS estimated at 15% to 25% 3 to 5 years posttransplant.9,10 Beyond this guiding principle, the optimal treatment of the majority of patients, both older and younger, with relapsed or refractory AML has lacked a uniform treatment strategy,11 despite the wide variety of regimens administered in numerous clinical trials carried out over >4 decades. However, recent regulatory agency approvals of a number of novel agents have provided the opportunity to revisit the approach to relapsed or refractory AML and may well pave the way for genuine progress in this historically difficult if not stagnant area. Here, we present illustrative cases that capture our treatment approach in the dynamic landscape of new diagnostic capabilities and in view of a burgeoning armamentarium of both approved and investigational therapies. We focus on treatment outside of a clinical trial because we recognize that clinical trials are not readily available in many parts of the world.
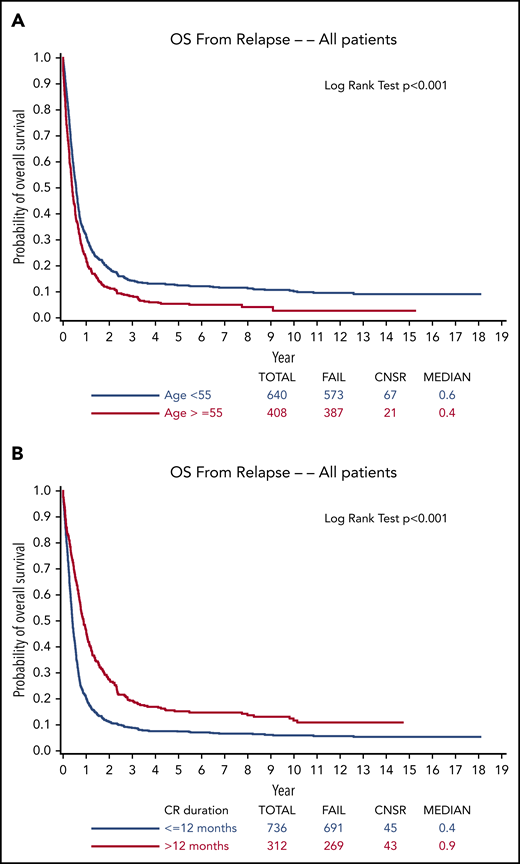
OS from relapse. Stratified by (A) age (<55 years or ≥55 years) and (B) duration of first CR (≤12 months or >12 months) from ECOG-ACRIN trials from 1984 to 2008.4 Reprinted from Ganzel et al4 with permission. CNSR, censor.
Definitions
Precise definitions of relapsed and refractory disease are important in the face of perpetually changing response criteria and in an effort to promulgate uniform terminology.12,13 There is no agreed-upon definition of primary refractory disease (PRD). Some consider PRD to mean failure after 1 cycle of induction chemotherapy. We consider PRD, also known as PIF, to be a state of persistent leukemia with >5% blasts after at least 2 cycles of intensive induction therapy, as put forward in the 2017 European LeukemiaNet (ELN) guidelines14 (Table 1). This 5% threshold, established decades ago and entrenched in daily practice, is arbitrary and could be revisited because we are now less dependent solely on morphology, with highly sensitive techniques for the detection of minimal/measurable residual disease (MRD). In the past, some argued that 1 cycle should include high-dose cytarabine (HiDAC), given the ability of HiDAC to overcome resistance to standard-dose cytarabine.15 In the current era with increasing use of nonintensive venetoclax-based regimens and targeted therapy, we speculate that this terminology requires further revision. Guided by recent prospective data,16,17 we tend to use hypomethylating agents (HMAs) or low-dose cytarabine (LoDAC) combined with venetoclax for 2 cycles before changing; however, some patients take longer to respond and, if tolerating therapy without obvious disease progression, we continue this regimen. For targeted therapy, median time to remission is typically over 2 months,6,18,19 patients can respond after even 4 to 6 months, and thus defining treatment failure is an even greater challenge. We generally continue until clear progressive disease.
Definitions of relapsed and refractory AML
| Category | Definition |
|---|---|
| PIF (PRD) | Lack of CR or CRi following at least 2 cycles of intensive chemotherapy |
| Hematologic relapse | Detection of ≥5% blasts in the BM, identification of circulating blasts, or emergence of extramedullary disease |
| MRD | Detection of MRD defined by molecular techniques or multiparameter flow cytometry after an MRD− CR |
| Category | Definition |
|---|---|
| PIF (PRD) | Lack of CR or CRi following at least 2 cycles of intensive chemotherapy |
| Hematologic relapse | Detection of ≥5% blasts in the BM, identification of circulating blasts, or emergence of extramedullary disease |
| MRD | Detection of MRD defined by molecular techniques or multiparameter flow cytometry after an MRD− CR |
Modified from the ELN guidelines in Döhner et al.14
BM, bone marrow; CR, complete remission; CRi, CR with incomplete hematologic recovery; MRD, minimal/measurable residual disease.
Relapsed disease falls into 2 main categories: clear morphologic relapse, which may be in the bone marrow or extramedullary, or the emergence of MRD after initial clearance (Table 1). The presence of 5% or greater leukemic blasts (acknowledging again that this percentage is arbitrary) in the bone marrow or detection of blasts in the peripheral blood fulfills criteria for morphologic relapse, unless another possible etiology to explain the blasts is identified, for example, as a result of robust hematologic reconstitution or stimulation of myeloid growth factors. In these settings, we repeat a bone marrow evaluation, even weekly if needed, to distinguish between recurrence and anticipated hematologic maturation; often molecular studies or the blast phenotype by flow cytometry can quickly differentiate the 2 processes. MRD describes a state in which the patient has evidence of leukemia detected by highly sensitive techniques such as real-time quantitative polymerase chain reaction, next-generation sequencing, or advanced flow cytometry without the presence of blasts sufficient to meet criteria for relapse.20-25 The definition is an evolving one based on available assays with varying thresholds of detection, detailed discussion of which is reviewed in Xiao et al.26 Although the detection of abnormal blasts by flow cytometry with a similar phenotype to the original disease raises strong suspicion for impending morphologic relapse, the prognostic value of some molecular studies is murkier and may vary by mutation. For some subtypes such as core-binding factor AML and nucleophosmin 1 (NPM1)-mutated AML, molecular MRD is associated with a higher relapse rate.27 Studies are needed to clarify technical aspects of MRD measurement and the clinical significance of the detection of abnormal clones, some of which may be preleukemic in nature and may not portend inevitable relapse.14,27
Prognostic factors
Historically, duration of first complete remission (CR) was a major factor for risk stratification in relapsed AML, with poorest outcomes seen in patients relapsing within the first year following initial induction therapy.28-30 Other independent factors that play a role in a patient’s clinical course include age, cytogenetics at diagnosis, certain molecular features such as FLT3–internal tandem duplication (ITD) mutation status, and history of prior allogeneic HCT.23,31-33 Two main prognostic indices were developed to stratify patient outcomes following relapse incorporating some of these variables: the European Prognostic Index (EPI) and the Groupe Ouest Est d’Etude des Leucemies et Autres Maladies du Sang (GOELAMS).2,34,35 An important caveat about these scoring systems is that, although they give a sense of a patient’s general prognosis regardless of risk stratification, the overall outcome is typically poor. Furthermore, these prognostic indices likely require revision in the era of advanced molecular studies and targeted agents. Therefore, in the hustle and bustle of a busy clinic day, we do not routinely rely on these scoring systems, but rather focus on the biology of a patient’s disease to guide therapy.
Patient 1
Early relapsed AML in a young patient
The first case is a 30-year-old otherwise healthy woman with AML and extramedullary disease at presentation who relapsed only 2 months after intensive induction and consolidation chemotherapy. She originally presented with cervical adenopathy and a white blood cell (WBC) count of 322 × 103/μL requiring emergent leukapheresis. Bone marrow studies revealed a hypercellular marrow with >90% abnormal myeloid blasts (CD34+, CD11b+, CD15+, CD33+, CD117+, CD123+, HLA-DR+) and further analyses identified a DNMT3A mutation and normal karyotype. Positron emission tomography–computed tomography detected fluorodeoxyglucose-avid nodes limited to the cervical and pelvic region; lumbar puncture with intrathecal chemotherapy confirmed no malignant cerebrospinal fluid cells. She received induction with cytarabine plus daunorubicin (90 mg/m2) and bone marrow 1 month after treatment demonstrated an MRD− CR both by flow cytometry and molecular studies. She elected not to pursue allogeneic HCT and received 4 cycles of consolidation therapy with HiDAC. However, 2 months after completing consolidation, her cervical adenopathy returned and bone marrow studies revealed 55% blasts with the same phenotype and mutation profile as her original disease. She was swiftly reinduced with MEC (mitoxantrone/etoposide/cytarabine (MEC), leading to a second morphologic CR with evidence of MRD by flow cytometry (1.8% of WBCs). She proceeded to allogeneic HCT from a matched unrelated donor with an ablative conditioning regimen and remains in CR 1 year after transplant without MRD. Nevertheless, her long-term prognosis is precarious, given that she underwent transplant with MRD36,37 together with the short duration of the first CR.
The field is rapidly moving away from indiscriminate multiagent conventional cytotoxic chemotherapy. However, in this setting of early relapse in a young patient whose leukemia cells do not express a targetable mutation, in whom cure with allogeneic HCT is the clear goal, we pursue an initial approach with intensive chemotherapy. Individual institutions and investigators often have a preferred chemotherapy regimen for reinduction therapy, but one cannot be dogmatic in the absence of randomized trials. Early studies supporting the use of HiDAC emerged in the late 1980s.15,38 However, 2 other regimens we frequently consider are MEC39 and fludarabine/cytarabine/granulocyte colony-stimulating factor (G-CSF)/idarubicin (FLAG-IDA).40 Prior treatment history and side-effect profile guide our specific reinduction choice, but we use classical HiDAC (3 g/m2) less often than in the past in an effort to minimize cytarabine-related toxicities and prolonged cytopenias; we are motivated by studies in consolidation suggesting that lower doses of cytarabine (1.5-2 g/m2) appear equally effective and less toxic than classical HiDAC.41
The efficacy of these regimens and similar variations is consistently relatively disappointing with CR rates ranging from ∼20% to 65% with a median duration of response typically <1 year.40,42-50 Although there is no proof that such high- or intermediate-dose regimens are better than repeating standard-dose induction among previously untreated patients if the day 14 marrow shows persistent disease, data from the German AML Cooperative Group suggest that patients with poor-risk disease such as those with >40% blasts on day 16 marrow fare better with more intensive cytarabine-containing regimens.51 Some groups prefer regimens with cladribine (cladribine/arabinoside cytosine/mitoxantrone/G-CSF)52,53 or clofarabine (clofarabine/cytarabine/G-CSF).54,55 However, these regimens have yielded similarly relatively poor outcomes despite their initial promise.
Implications of a FLT3 mutation
A somatic FLT3 mutation is identified in nearly 30% of AML patients and the development of several small molecule inhibitors of FLT3 has created a new treatment paradigm (Table 2).6,56-59 When in a patient’s disease course to administer these targeted agents is an area of ongoing investigation, but 1 important randomized trial has contributed to a change in the standard of care for relapsed or refractory patients with FLT3-ITD and/or FLT3–tyrosine kinase domain mutations. Recent data from the ADMIRAL study, a phase 3 trial investigating the potent FLT3 inhibitor gilteritinib in primary refractory or first relapse FLT3-mutated (FLT3-ITD and FLT3-tyrosine kinase domain D835 or I836) disease, not only led to the approval of this agent by the US Food and Drug Administration (FDA) in November 2018 for relapsed or refractory FLT3-mutated disease, but also has led to changes in treatment strategies.6 Gilteritinib monotherapy resulted in a median OS of 9.3 months compared with 5.6 months in the reinduction chemotherapy (low-dose cytarabine, azacitidine, MEC, or FLAG-IDA) arm. Two subgroups found to have longer survival were patients with high FLT3-ITD allelic ratios (median OS, 7.1 months compared with 4.3 months) and those with certain comutations, such as NPM1 and DNMT3A (median OS, 10.8 months vs 8.9 months).
New targeted therapeutics in current clinical use for relapsed or refractory AML
| Agent | Target | CR+CRh/CRi/CRp % (CR %) | Median survival, mo | Approved population | FDA-approval status | Reference for FDA-approved indication and support for use in the unapproved setting |
|---|---|---|---|---|---|---|
| Gilteritinib | FLT3 | 34 (21.1) | 9.3 | FLT3-mutated R/R AML | 2017 | 6 |
| Enasidenib | IDH2 | 26.6 (20.2) | 9.3 | IDH2-mutated R/R AML | 2017 | 18 |
| Ivosidenib | IDH1 | 30.4 (21.6) | 8.8 | IDH1-mutated R/R or untreated AML | 2018 | 19 |
| Gemtuzumab ozogamicin | CD33 | 33 (26) | 8.4 | CD33+ untreated or R/R AML in adults or pediatric patients 2 y or older | 2017 | 101 |
| HMA/LoDAC + venetoclax | BCL-2 | 67 (54)16 ; 54 (26)17 (untreated) | 17.516 ; 10.117 (untreated) | Untreated AML in patients 75 y and older unfit for chemotherapy | 2018 | 16,17,68-74* |
| Agent | Target | CR+CRh/CRi/CRp % (CR %) | Median survival, mo | Approved population | FDA-approval status | Reference for FDA-approved indication and support for use in the unapproved setting |
|---|---|---|---|---|---|---|
| Gilteritinib | FLT3 | 34 (21.1) | 9.3 | FLT3-mutated R/R AML | 2017 | 6 |
| Enasidenib | IDH2 | 26.6 (20.2) | 9.3 | IDH2-mutated R/R AML | 2017 | 18 |
| Ivosidenib | IDH1 | 30.4 (21.6) | 8.8 | IDH1-mutated R/R or untreated AML | 2018 | 19 |
| Gemtuzumab ozogamicin | CD33 | 33 (26) | 8.4 | CD33+ untreated or R/R AML in adults or pediatric patients 2 y or older | 2017 | 101 |
| HMA/LoDAC + venetoclax | BCL-2 | 67 (54)16 ; 54 (26)17 (untreated) | 17.516 ; 10.117 (untreated) | Untreated AML in patients 75 y and older unfit for chemotherapy | 2018 | 16,17,68-74* |
BCL-2, B-cell lymphoma 2; CRh, CR with partial hematological recovery; CRp, CR with incomplete platelet recovery; IDH, isocitrate dehydrogenase; R/R, relapsed or refractory. See Table 1 for expansion of other abbreviations.
Selected additional recent publications and abstracts.
How would we have treated this patient if her disease had been FLT3-ITD+? Occasionally, patients with highly proliferative disease may require urgent cytoreduction with cytotoxic chemotherapy. Guided by the ADMIRAL study, we now strongly consider gilteritinib if the patient has FLT3-mutated AML in first relapse rather than intensive chemotherapy, taking into account the patient’s clinical course and disease biology. We evaluate for loss or gain of targetable mutations in all patients with relapse. The poor survival rate in either group in the ADMIRAL study is conspicuous. Whether FLT3 inhibitors with chemotherapy will be more effective than FLT3 inhibitors alone will be determined in future randomized clinical trials. Additionally, several studies suggest a benefit for FLT3 inhibitors after allogeneic HCT as maintenance.6,58,60-63 Based on these data, we would have encouraged such a strategy following transplant if our patient had a FLT3-ITD mutation. An ongoing randomized trial will further clarify the role of FLT3 inhibition after allogeneic HCT.64
Patient 2
PIF in a patient with AML
A 52-year-old man without significant medical history presented with shortness of breath and was found to be anemic with a hemoglobin of 6.2 g/dL with blasts identified in the peripheral blood. A bone marrow biopsy established a diagnosis of AML with inversion(3) by cytogenetics [46,XY,inv(3)(q21q26)] without other associated mutations, a karyotype associated with a particularly poor prognosis. He initially received cytarabine-plus-daunorubicin induction with persistence of over 70% blasts. Then he received classical HiDAC, but had 33% blasts on follow-up marrow evaluation. After failing a clinical trial, he received reinduction with MEC followed again by progression of disease. Finally, 8 months after diagnosis, he received 2 cycles of azacitidine and venetoclax, as this regimen was becoming available, and achieved a morphologic CR, though with MRD by flow cytometry. He proceeded to allogeneic HCT and remained disease-free for over 2 years.
This patient falls into the category of PIF having failed to achieve CR after at least 2 courses of intensive chemotherapy. The emergence of regimens combining the oral B-cell lymphoma 2 (BCL-2) inhibitor venetoclax with HMAs (decitabine or azacitidine) or LoDAC is rapidly changing our treatment approach (Table 2).16,17,65 If we were treating this patient today, this regimen would have been our first choice for PIF, outside of a trial. Although mechanistically much remains to be learned about this regimen,66 results from untreated patients are transformative and have been met with unbridled euphoria. Initial studies with single-agent venetoclax revealed a response rate of only 19% in patients with relapsed or refractory disease.67 However, when combined with HMAs in older adults with newly diagnosed AML not eligible for standard induction therapy, remarkable apparent synergism resulted in a response rate of over 70%.16,65 Furthermore, the regimen is typically well tolerated, is not limited to patients with a specific mutation, and often can be given outpatient as we do in our practice usually after the first cycle. Studies are limited in the relapsed or refractory disease setting and are primarily retrospective in nature, though initial clinical experience suggests promising results. However, the variability of reported outcomes in early studies is unsettling and may be due to the small numbers of patients: published CR/CR with incomplete hematologic recovery (CRi) rates range from 12% to 51%.68-74
One of the features of HMA plus venetoclax is the relatively rapid clinical response with maximum responses achieved after only 1 to 2 cycles,16,65 which is an important distinction from single-agent HMA for which responses may take several months of therapy.75 We typically perform a bone marrow evaluation after the first cycle, particularly if the patient remains cytopenic, to determine whether the cytopenias are attributable to persistent disease or drug-induced aplasia. In some patients, we reduce the duration of venetoclax dosing from 28 days to 21 or even 14 days because of prolonged count recovery.
In eligible patients, we encourage allogeneic HCT after 1 to 2 cycles of therapy, ideally when morphologic CR is achieved. We still pursue transplantation in appropriate patients even with MRD36,37 and/or incomplete count recovery in the relapsed or refractory setting if it is unlikely that the patient can be rendered MRD− and in light of the potential of transplant to cure these patients. If transplant is not the intended course, we continue HMA plus venetoclax indefinitely, as long as the patient has a continued response and acceptable side-effect profile. The optimal duration of therapy remains to be elucidated. That patients survive long enough to ask about the optimal duration of treatment is a victory in itself.
A randomized trial is needed to determine whether, even for older newly diagnosed patients fit for intensive reinduction chemotherapy, a venetoclax-based approach is the most effective, particularly if future studies determine that co-occurring mutations such as isocitrate dehydrogenase 1/2 (IDH1/2) and NPM1 predict response, as is becoming increasingly evident.17,68 Since the combination of HMA or LoDAC plus venetoclax burst onto the scene, it has been rapidly and enthusiastically embraced not only for untreated patients in particular, but also for those with relapsed or refractory disease without evidence from prospective studies. Although we are optimistic about this regimen, we are highly supportive of clinical studies from which we can learn about venetoclax-based approaches, including in combination with chemotherapy76,77 or other novel agents.
Relapse following allogeneic HCT
What if this patient ultimately relapses after undergoing transplantation? The lack of options for relapse following transplantation is well recognized and clinical trials are typically our preferred approach, unless the patient has a targetable mutation. In patients with CD33+ disease, gemtuzumab is an option, but there is a risk of veno-occlusive disease, also called sinusoidal obstructive syndrome, particularly if administered in close proximity (within 3 months) to allogeneic HCT (Table 2); therefore, we tend to prefer other therapies, such as targeted agents, if suitable. Other options we consider are HMAs,78-80 cellular therapies such as donor lymphocyte infusion,81 a combination of both,82-84 or a second transplant from a new donor in appropriate patients in second CR.10 The latter approaches and other investigational cellular therapies are guided by our transplant colleagues.
Patient 3
Relapsed IDH+ AML in the older patient
An 86-year-old woman was originally treated with azacitidine for AML diagnosed as part of an evaluation of pancytopenia requiring red blood cell transfusions. At diagnosis, a bone marrow biopsy identified 64% blasts with cytogenetics notable for a 5q deletion. Molecular studies were unrevealing. She enjoyed 2 years of excellent response with an improvement in her peripheral blood counts until a bone marrow evaluation detected 30% to 40% blasts along with worsening pancytopenia. A complete molecular evaluation led to discovery of an IDH1 mutation, prompting the initiation of ivosidenib, which had been recently approved. Within weeks her peripheral blood counts began to improve. She continues without evidence of recurrent disease on ivosidenib for over a year following relapse.
Relapsed AML in the older patient presents a distinct challenge as the fitness and wellness of the patient plays as important a role as the disease status.85 Clinical trial options are frequently limited. We typically avoid intensive chemotherapy. Although we do not impose strict age limits for specific treatment approaches and take into account functional independence, emotional support, and overall goals of treatment, we have great respect for octogenarians.
This case underscores the importance of a complete molecular investigation at the time of relapse. We always check for IDH and FLT3 mutation status. We also perform a more comprehensive panel of mutations by next-generation sequencing, primarily to evaluate for potential clinical trials. The principle of clonal evolution in AML is now well established86,87 and, in the era of targeted inhibitors, a first step in all relapsed patients is to carry out molecular studies. Ivosidenib and enasidenib are novel oral agents targeting IDH1 and IDH2, respectively, and are specifically included in the 2019 National Comprehensive Cancer Network (NCCN) guidelines for relapsed or refractory patients.11,18,19,88 The phase 1 study published in 2018 leading to ivosidenib’s FDA approval identified a CR rate among relapsed or refractory patients of 21.6% with a median CR duration of 9.3 months.19 However, long-term data are currently limited and no randomized data have been presented.
The outcome for patients with IDH2 mutations treated with enasidenib is similarly encouraging (Table 2).18 Both enasidenib and ivosidenib are associated with a risk of IDH differentiation syndrome, a systemic cardiorespiratory distress syndrome associated with the differentiation of immature leukemic cells into a mature phenotype reminiscent of the differentiation syndrome observed in some patients with APL.89 Symptoms are nonspecific, but include unexplained fever, volume overload, and multisystem organ dysfunction. Steroids are typically effective. We temporarily interrupt targeted therapy if the presentation requires hospitalization or if severe symptoms persist for >48 hours after steroids have been started; we worry less about holding treatment than we do in APL.18,19,90,91
Our patient tolerated ivosidenib without side effects at a dose of 500 mg daily. She has monthly electrocardiograms as QT-interval prolongation was seen in 24.6% of patients in the original study.19 Although in many patients we pursue regular bone marrow biopsies for disease assessment, for older patients we try to minimize invasive procedures. With normal blood counts and no detectable disease in the peripheral blood, we monitor patients with only clinical evaluations and complete blood counts. Because the median time to CR is over 2 months for ivosidenib19 and enasidenib,18 we continue treatment as long as patients appear to be deriving some clinical benefit before changing our approach. Stein et al reported that 30% of patients with stable disease after 90 days on enasidenib subsequently had a response.88 In many parts of the world, IDH inhibitors are not available. Therefore, for IDH-mutated relapsed or refractory patients, we consider HMA or LoDAC plus venetoclax. If venetoclax is not available, we would consider HMA alone or best supportive care.
Not only have the IDH and FLT3 inhibitors already transformed the treatment of AML, but there are also many more targeted approaches in development, either alone or in combination with HMA or chemotherapy. Approved in Japan, quizartinib is a potent FLT3 inhibitor found to be superior as a single agent to chemotherapy in the phase 3 QuANTUM-R trial in the relapsed or refractory setting.58 In addition to the trove of promising targeted agents for relapsed or refractory AML, diverse immunotherapies are also under investigation, ranging from checkpoint inhibitors to bispecific T-cell engagers to antibody-drug conjugates.
Resistance to targeted therapy
Although our patient responded well to ivosidenib, her disease may at some point develop resistance. Detailed investigations have identified new IDH mutations in some patients on IDH-directed therapy, eliminating susceptibility of the disease to inhibitors.92 Furthermore, second-site mutations may occur in trans from the original IDH mutation, which is a novel resistance mechanism. In some patients, disease resistance results from progression of preexisting non-IDH–mutated clones that emerge under the selective pressure of anti-IDH therapy. There are even reports of IDH1 mutations undetected at diagnosis that arise in patients with IDH2-mutated disease receiving enasidenib.93,94 Secondary mutations in FLT3 or other key signaling pathways such as RAS/MAPK have been identified as 1 resistance mechanism to FLT3 inhibitors.95-97 Furthermore, not all clones in a patient’s relapsed disease may harbor a given mutation, limiting the efficacy of targeted treatment. Combining targeted therapy with other agents such as HMAs may be a powerful approach to overcome some disease resistance.98,99
Conclusions
If you have built castles in the air, your work need not be lost; that is where they should be.
Now put the foundations under them.
—Henry David Thoreau
Historically, effective treatment of relapsed or refractory AML has been like Thoreau’s castle in the air, the material of dreams for hematologists. With molecular testing, targeted agents, and a myriad of novel treatment combinations, we find ourselves in a new era of medicine. A schematic capturing our current approach with factors guiding our decision process is presented in Figure 2. For young and/or fit patients in first relapse after >6 months in CR,2 we treat with cytotoxic chemotherapy; however, we favor gilteritinib if the patient has an FLT3-ITD mutation. We recognize that this 6-month time period is arbitrary. If relapse occurs <6 months from CR, our approach depends on the biology of the disease: if adverse cytogenetic or molecular abnormalities are present, we treat with an inhibitor if a mutation is present or HMA/LoDAC plus venetoclax if no mutation is present, rather than cytotoxic chemotherapy. If the patient is older, less fit, has PIF, or multiple relapses, we treat with HMA/LoDAC plus venetoclax, unless the patient has a targetable mutation, in which case we treat with the appropriate inhibitor.
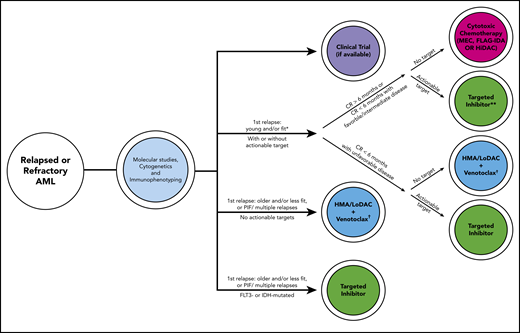
Schematic capturing our current general approach for relapsed or refractory patients with AML with some factors guiding the clinical decision process. Risk stratification by 2017 ELN criteria.14 Approved targeted inhibitors include gilteritinib (FLT3), ivosidenib (IDH1), enasidenib (IDH2). *Assuming patient has already received intensive consolidation. **Randomized phase 3 data are only currently available for gilteritinib,6 not ivosidenib or enasidenib. †HMA alone if venetoclax unavailable. FLAG-IDA, fludarabine/cytarabine/granulocyte colony-stimulating factor/idarubicin; HiDAC, high-dose cytarabine; HMA, hypomethylating agent; IDH, isocitrate dehydrogenase; LoDAC, low-dose cytarabine; MEC, mitoxantrone/etoposide/cytarabine; PIF, primary induction failure.
Schematic capturing our current general approach for relapsed or refractory patients with AML with some factors guiding the clinical decision process. Risk stratification by 2017 ELN criteria.14 Approved targeted inhibitors include gilteritinib (FLT3), ivosidenib (IDH1), enasidenib (IDH2). *Assuming patient has already received intensive consolidation. **Randomized phase 3 data are only currently available for gilteritinib,6 not ivosidenib or enasidenib. †HMA alone if venetoclax unavailable. FLAG-IDA, fludarabine/cytarabine/granulocyte colony-stimulating factor/idarubicin; HiDAC, high-dose cytarabine; HMA, hypomethylating agent; IDH, isocitrate dehydrogenase; LoDAC, low-dose cytarabine; MEC, mitoxantrone/etoposide/cytarabine; PIF, primary induction failure.
Final thoughts
Propelled by effective oral targeted agents, we are particularly enthusiastic about therapeutic strategies that avoid intensive chemotherapy and prolonged inpatient admissions in an effort to improve overall quality of life (Table 3). Although there may be a rare patient for whom 3 or even more courses of intensive chemotherapy may be effective, such treatment is no longer the only option and is generally not advised.100 Many advances have occurred since the last “How I treat refractory and early relapsed AML” was published in 2015. We anticipate that the next such publication will address more new targets, more novel targeted agents, and combinations of targeted agents with each other (doublets, triplets, and beyond) and with chemotherapy. The identification of MRD by increasingly sensitive and widely available techniques will likely establish a more definitive role for MRD in guiding therapy. Our major focus should be on prevention of recurrent disease. Indeed, we predict that there will be fewer patients with relapsed or refractory disease as more patients are likely to be cured with initial therapy.
Our central guiding principles in the setting of relapsed or refractory AML
| Guiding principles for relapsed or refractory AML |
|---|
| 1. Enroll patients on clinical trials, when available |
| 2. Perform molecular testing to identify potential for targeted therapy and drug development |
| 3. Decipher mechanisms of treatment resistance |
| 4. Avoid reinduction attempts with multiple sequential courses of intensive cytotoxic chemotherapy |
| 5. Ensure appropriate patients proceed to allogeneic HCT, ideally when patients are MRD− |
| Guiding principles for relapsed or refractory AML |
|---|
| 1. Enroll patients on clinical trials, when available |
| 2. Perform molecular testing to identify potential for targeted therapy and drug development |
| 3. Decipher mechanisms of treatment resistance |
| 4. Avoid reinduction attempts with multiple sequential courses of intensive cytotoxic chemotherapy |
| 5. Ensure appropriate patients proceed to allogeneic HCT, ideally when patients are MRD− |
Acknowledgments
The authors thank Bernadette Cuello and Suzanne Moshett for assistance with preparation of the manuscript, and Susan Weil for assistance with preparation of figures. The authors also thank Alexander E. Perl and Jacob M. Rowe for helpful comments on the manuscript.
S.D. was supported in part by a Clinical Scholars 2T32 CA009512-31 through Memorial Sloan Kettering internal funding.
Authorship
Contribution: S.D. and M.S.T. wrote and edited the manuscript.
Conflict-of-interest disclosure: M.S.T. receives research funding from AbbVie, Cellerant, Orsenix, ADC Therapeutics, Biosight, Amgen, Rafael, and Glycomimetics; participates on the following advisory boards: AbbVie, BioLineRx, Daiichi-Sankyo, Orsenix, KAHR, Rigel, Nohla, Delta Fly Pharma, Tetraphase, Oncolyze, Jazz Pharma, Roche, Biosight, and Novartis; and receives royalties from UpToDate. S.D. declares no competing financial interests.
Correspondence: Martin S. Tallman, Leukemia Service, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: tallmanm@mskcc.org.
