


Abstract
Adoptive immunotherapy using B-cell–targeted chimeric antigen receptor (CAR)-modified T cells to treat hematologic malignancies is transforming cancer care for patients with refractory or relapsed diseases. Recent and anticipated regulatory approval for products targeting acute lymphoblastic leukemia, lymphomas, and multiple myeloma have led to global implementation of these novel treatments. The rapidity of commercial utilization of CAR–T-cell therapy has created a largely unexplored gap in patient supportive-care approaches. Such approaches are critical in these complex patients given their high net state of immunosuppression prior to CAR–T-cell infusion coupled with unique acute and persistent insults to their immune function after CAR–T-cell infusion. In this “How I Treat” article, we focus on key questions that arise during 3 phases of management for patients receiving CD19-targeted CAR-T cells: pre CAR–T-cell infusion, immediate post CAR–T-cell infusion, and long-term follow-up. A longitudinal patient case is presented for each phase to highlight fundamental issues including infectious diseases screening, antimicrobial prophylaxis, immunoglobulin supplementation, risk factors for infection, and vaccination. We hope this discussion will provide a framework for institutions and health care providers to formulate their own approach to preventing infections in light of the paucity of data specific to this treatment modality.
Introduction
Adoptive immunotherapy using B-cell–targeted chimeric antigen receptor (CAR)-modified T (CAR-T) cells to treat hematologic malignancies has fundamentally transformed cancer treatment paradigms. This novel therapy for patients with B-cell malignancies, including acute lymphoblastic leukemia (ALL) and large B-cell lymphomas, has had unprecedented success.1-6 There has been rapid and wide-scale utilization of these treatments since the commercial approval of the first 2 CD19-targeted CAR–T-cell products by the US Food and Drug Administration in 2017: (1) tisagenlecleucel (Kymriah; Novartis) for refractory or relapsed (R/R) B-cell ALL in patients aged 0 to 25 years and R/R lymphomas in adults and (2) axicabtagene ciloleucel (Yescarta; Kite/Gilead) for R/R lymphomas in adults. Additionally, there are hundreds of ongoing clinical trials around the world.
Although CD19-targeted CAR–T-cell therapies are promising treatment options, the currently approved products have serious toxicities including cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS).4,6-14 These acute toxicities and their treatment add to the already high net state of immunosuppression and infection risk due to patients’ underlying malignancy, prior cytotoxic treatments, and pre CAR–T-cell infusion lymphodepletion chemotherapy. Prolonged cytopenias, especially due to the “on-target, off-tumor” depletion of normal CD19-expressing B cells (Figure 1), may result in profound and prolonged immune deficits given that CAR-T cells are a living drug that may persist for years.4,11,15-21 Thus, CAR–T-cell immunotherapies pose unique challenges for acute and long-term infection prevention. The rapidity of commercialization and implementation of CD19-targeted CAR–T-cell immunotherapies has created a largely unexplored gap in the supportive-care approaches to maintaining not only cancer-free, but also infection-free, survival.
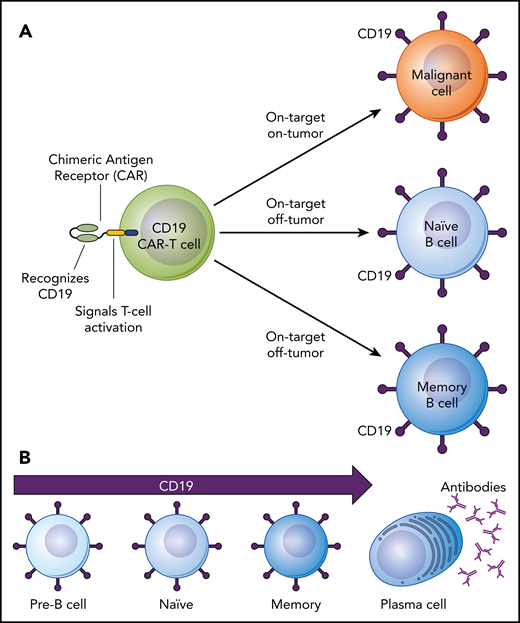
On-target, off-tumor side effects of CD19-targeted CAR–T-cell therapy. (A) Depiction of a CD19-targeted CAR-T cell that has both “on-target, on-tumor” and “on-target, off-tumor” activity. (B) The lineage of B cells from early to fully differentiated cells depicting expression of the CD19 cell surface antigen on pre-B cells, naïve B cells, and memory B cells but not on antibody-producing plasma cells.
On-target, off-tumor side effects of CD19-targeted CAR–T-cell therapy. (A) Depiction of a CD19-targeted CAR-T cell that has both “on-target, on-tumor” and “on-target, off-tumor” activity. (B) The lineage of B cells from early to fully differentiated cells depicting expression of the CD19 cell surface antigen on pre-B cells, naïve B cells, and memory B cells but not on antibody-producing plasma cells.
This “How I Treat” article arose from a need for pragmatic standards for infection prevention in patients treated with CD19-targeted CAR-T cells, while recognizing the dearth of data specific to this treatment modality. We present a longitudinal patient case to facilitate a discussion of how we approach infection screening, monitoring, prophylaxis, and vaccination in CD19-targeted CAR–T-cell recipients. The discussion is organized into frequently asked questions (FAQs) to facilitate good clinical practice. Suggested medication doses are based on adults. Supplemental FAQs (sFAQs) are available on the Blood Web site. None of the recommendations are based on randomized, controlled clinical trials in this patient population; instead, they are based on our expert opinion, opinions of others in the field, and approaches used in other relevant contexts. Many of these recommendations align with generally accepted infection-prevention strategies in patients with leukemias or lymphomas receiving high-dose corticosteroids, B-cell–targeted therapies like rituximab, or hematopoietic cell transplantation (HCT). The recommendations are intended as general guidance for management of patients receiving CD19-targeted CAR-T cells.
Infectious diseases screening prior to CAR–T-cell therapy
Clinical case
A 52-year-old woman diagnosed with diffuse large B-cell lymphoma had persistent disease despite 4 prior treatment regimens including an anti-CD20 antibody, alkylating agents, and an anthracycline. She had no other notable past medical history and had high performance status (Eastern Cooperative Oncology Group [ECOG] score of 1) and normal organ function including normal left-ventricular ejection fraction. She was not a suitable candidate for an HCT due to refractory disease and was referred for treatment with CD19-targeted CAR–T-cell therapy using axicabtagene ciloleucel.
FAQs
FAQ 1: What screening tests should be performed for infectious diseases prior to CD19-targeted CAR–T-cell therapy?
At a minimum, we recommend screening all patients for HIV antibodies, hepatitis B virus (HBV) surface antigen (HBsAg), HBV surface antibody (anti-HBs), HBV core antibody (anti-HBc), and hepatitis C virus (HCV) antibody with reflex nucleic acid testing if any of these tests are positive (Table 1). Additional serologic screening can be considered for herpes simplex virus 1/2 (HSV1/2) and varicella-zoster virus (VZV), especially for patients not receiving acyclovir prophylaxis; cytomegalovirus (CMV); human T-cell lymphotropic virus type 1 (HTLV-1); Treponema pallidum (syphilis); and Toxoplasma gondii. Given that there is no reliable way to predict which patients will require high-dose corticosteroids for CRS and/or ICANS, which may increase risk for reactivation of latent infections, screening for Mycobacterium tuberculosis should be considered in patients with risk factors for exposure; testing may be indeterminate in patients with severe lymphopenia. In addition to the increased risk for reactivation of M tuberculosis in cancer patients,22 CD19-targeted CAR–T-cell therapy recipients may require treatment with tocilizumab, an interleukin 6 (IL-6) receptor antagonist, which is independently associated with increased risk for active M tuberculosis infections.23 Similarly, screening for antibodies to Strongyloides stercoralis, or empiric treatment with ivermectin, should be considered in patients with risk factors for infection (eg, extended time spent in tropical or subtropical regions). Screening approaches will be guided by established and evolving local and state regulatory requirements.
Infectious diseases screening prior to CD19-targeted CAR–T-cell therapy
| Screening for infectious diseases |
|---|
| Required |
| HIV using the fourth-generation antigen/antibody combination HIV-1/2 immunoassay* |
| HBsAg, anti-HBs, and anti-HBc* |
| HCV IgG* |
| Consider† |
| HSV-1 and HSV-2 IgG‡ |
| VZV IgG |
| CMV IgG |
| HTLV-1 IgG |
| Toxoplasma gondii IgG |
| Treponema pallidum (syphilis) treponemal or nontreponemal test |
| M tuberculosis skin test and/or blood interferon-γ release assay§ |
| S stercoralis IgG or empiric treatment§ |
| Screening for infectious diseases |
|---|
| Required |
| HIV using the fourth-generation antigen/antibody combination HIV-1/2 immunoassay* |
| HBsAg, anti-HBs, and anti-HBc* |
| HCV IgG* |
| Consider† |
| HSV-1 and HSV-2 IgG‡ |
| VZV IgG |
| CMV IgG |
| HTLV-1 IgG |
| Toxoplasma gondii IgG |
| Treponema pallidum (syphilis) treponemal or nontreponemal test |
| M tuberculosis skin test and/or blood interferon-γ release assay§ |
| S stercoralis IgG or empiric treatment§ |
If positive, perform reflex nucleic acid testing.
Screening for these pathogens may impact antimicrobial prophylaxis strategies and are routinely performed in the context of donor and recipient evaluations for HCT or solid organ transplantation.35,74 Additional risk factors in CD19-targeted CAR–T-cell therapy recipients, such as high-dose corticosteroids and tocilizumab for CRS or ICANS, may increase risk for reactivation of latent infections such as M tuberculosis and S stercoralis. Given that latent infection may be asymptomatic, and symptoms of reactivation are often nonspecific, screening may improve risk stratification and identification of infections after CAR–T-cell therapy.
Patients not already receiving antiviral prophylaxis for herpesviruses should have serologic screening; if positive, they should be started on antiviral prophylaxis as per FAQ 4.
In patients with risk factors for exposure.
FAQ 2: Can CD19-targeted CAR–T-cell therapy be administered in patients with ongoing or new infections?
Administration of lymphodepleting chemotherapy and CAR–T-cell infusion should generally be delayed, when feasible, in patients with serious, uncontrolled infections. One study demonstrated that ∼10% of severe or life-threatening infections progressed from infections present prior to CAR–T-cell infusion.7 Whether preexisting infection increases risk for severe CRS requires more study.7 All patients with respiratory symptoms should undergo workup consisting of multiplex polymerase chain reaction testing for respiratory viruses from the upper respiratory tract with consideration for chest radiographic imaging. In patients with upper and/or lower respiratory viral infection, a delay in CAR–T-cell therapy should be considered until resolution of symptoms, particularly for respiratory syncytial virus, parainfluenza viruses 1 to 4, influenza virus A or B, metapneumovirus, and adenovirus. Although delaying CAR–T-cell therapy until resolution of infections may be preferred, this must be balanced against the risk of malignancy progression.
Prophylaxis and monitoring for infectious diseases
Clinical case continued
The patient discussed in the clinical case was afebrile and clinically stable at her arrival evaluation. Her laboratory results were notable for a total serum immunoglobulin G (IgG) concentration of 350 mg/dL and an absolute neutrophil count of 1300 cells per mm3. She was given 1 dose of IV IgG (IVIG) at 400 mg/kg. The patient underwent leukapheresis to generate an autologous CD19-targeted CAR–T-cell product. Fifteen days later, she received conditioning chemotherapy consisting of fludarabine 30 mg/m2 per day and cyclophosphamide 500 mg/m2 per day on days −5, −4, and −3 before the administration of a single IV infusion of CAR-T cells at a target dose of 2 × 106 cells per kilogram on day 0. Valacyclovir, 500 mg orally twice daily, was started with the chemotherapy. The patient developed severe neutropenia with an absolute neutrophil count (ANC) of <0.5 × 109/L on day 1 post CAR–T-cell infusion prompting prophylactic levofloxacin (750 mg orally daily) and fluconazole (200 mg orally daily). On day 2, she developed tachycardia and fever with a temperature of 39.1°C and was transitioned from levofloxacin to cefepime 2 g every 8 hours IV. On day 3, she had continued fevers along with new hypotension that did not respond to fluids and required initiation of 2 vasopressors. She received a dose of tocilizumab 8 mg/kg IV followed by dexamethasone 10 mg IV. Given persistent symptoms and the development of mild hypoxemia, she received another dose of tocilizumab on days 4 and 5. Dexamethasone was continued every 6 hours through day 7. Blood cultures on day 6 were positive for Streptococcus mitis, and vancomycin 15 mg/kg IV every 12 hours was added. Due to the use of high-dose steroids, fluconazole was changed to posaconazole 300 mg once daily for prophylaxis with a mold-active agent. Her symptoms improved on day 7 and she began a steroid taper. Her ANC rose above 0.5 × 109/L on day 13.
FAQs
FAQ 3: What antibacterial prophylaxis should be used after CD19-targeted CAR–T-cell therapy?
The role of antibacterial prophylaxis for patients undergoing CD19-targeted CAR–T-cell therapy is not clear. Some centers implement fluoroquinolone prophylaxis during periods of severe neutropenia (ANC <0.5 × 109/L; Figure 2), whereas other centers may not routinely use antibiotic prophylaxis unless the neutropenic patient is in the outpatient setting.7,14 Additional considerations, including the use of myeloid growth factors, are discussed in sFAQ 1.
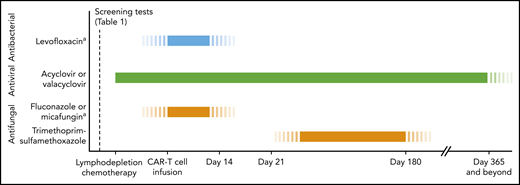
Timeline for antimicrobial prophylaxis in patients receiving CD19-targeted CAR–T-cell therapy. In patients meeting high-risk criteria, refer to Table 2 for additional recommendations for antimicrobial management and infection monitoring. Monitoring absolute CD4 T-cell counts can be considered for guidance regarding when to stop prophylaxis for herpesviruses and Pneumocystis jirovecii pneumonia (PCP), although the utility of this approach in cancer treatment–induced cytopenias is unclear. aDuring periods of severe neutropenia (ANC <0.5 × 109/L).
Timeline for antimicrobial prophylaxis in patients receiving CD19-targeted CAR–T-cell therapy. In patients meeting high-risk criteria, refer to Table 2 for additional recommendations for antimicrobial management and infection monitoring. Monitoring absolute CD4 T-cell counts can be considered for guidance regarding when to stop prophylaxis for herpesviruses and Pneumocystis jirovecii pneumonia (PCP), although the utility of this approach in cancer treatment–induced cytopenias is unclear. aDuring periods of severe neutropenia (ANC <0.5 × 109/L).
FAQ 4: What antiviral prophylaxis and monitoring strategies should be used for herpesviruses after CD19-targeted CAR–T-cell therapy?
We recommend prophylactic acyclovir 400 mg orally twice daily, or valacyclovir 500 mg orally twice daily, starting with lymphodepleting chemotherapy prior to CAR–T-cell infusion and continuing for at least 6 months post CAR–T-cell therapy (Figure 2). Although the data are limited, other herpesviruses such as CMV, Epstein-Barr virus (EBV), and human herpesvirus 6B (HHV-6B) do not appear to be frequent opportunistic infections after CD19-targeted CAR–T-cell therapy to date.7,14 We do not recommend routine serum or plasma nucleic acid test monitoring for herpesviruses. Targeted testing should be considered in the appropriate clinical context, especially in patients who received an HCT within the prior year or who require substantial immunosuppression for acute management after CD19-targeted CAR–T-cell therapy (see FAQ 13 and Table 2).
Antimicrobial management and infection monitoring in patients with CRS and/or ICANS
| Management and monitoring |
|---|
| • Empiric broad-spectrum antibiotics according to fever and neutropenia guidelines* |
| • ID consultation should be obtained to guide escalation and de-escalation of antimicrobial therapy, particularly in high-risk patients† |
| High-risk patients are those who meet any of the below criteria |
| o Receiving >1 dose of tocilizumab |
| o Requiring >3 days of ≥10 mg dexamethasone per day within a 7-day period |
| o Receiving 1 or more doses of methylprednisolone ≥1 g per day |
| o Receiving second-line agents for management of CRS or ICANS (eg, anakinra, siltuximab) |
| • Antibiotic de-escalation should be addressed on a daily basis with consideration for the type of immunosuppressive therapies that have been administered. |
| • Consider weekly CMV monitoring with serum polymerase chain reaction testing in high-risk patients who are CMV seropositive‡ |
| • Consider using mold-active azole prophylaxis with posaconazole in high-risk patients§ |
| Management and monitoring |
|---|
| • Empiric broad-spectrum antibiotics according to fever and neutropenia guidelines* |
| • ID consultation should be obtained to guide escalation and de-escalation of antimicrobial therapy, particularly in high-risk patients† |
| High-risk patients are those who meet any of the below criteria |
| o Receiving >1 dose of tocilizumab |
| o Requiring >3 days of ≥10 mg dexamethasone per day within a 7-day period |
| o Receiving 1 or more doses of methylprednisolone ≥1 g per day |
| o Receiving second-line agents for management of CRS or ICANS (eg, anakinra, siltuximab) |
| • Antibiotic de-escalation should be addressed on a daily basis with consideration for the type of immunosuppressive therapies that have been administered. |
| • Consider weekly CMV monitoring with serum polymerase chain reaction testing in high-risk patients who are CMV seropositive‡ |
| • Consider using mold-active azole prophylaxis with posaconazole in high-risk patients§ |
For institutions that use cefepime as a first-ine antibiotic, alternatives with a similar spectrum (eg, piperacillin-tazobactam or ceftazidime plus vancomycin) can be considered in select cases in discussion with the ID consult service given that cefepime is independently associated with neurotoxicity in the context of older age and acute kidney injury.75
Because patients will differ in regard to their exposure history, antimicrobial prophylaxis, treatment of prior/concomitant infections, and other factors, the ID service should be consulted to help guide management of these highly complex patients who will benefit from interdisciplinary care.
Consider for 1 mo following the last dose of immunosuppressive therapies, but duration may be modified according to a patient’s clinical course. Consider preemptive therapy at CMV viral load thresholds according to institutional guidelines for autologous HCT recipients. The cost-effectiveness and clinical utility of this approach are unknown at this time and should be refined when more data are available.
Consider for 1 mo following the last dose of immunosuppressive therapies, but duration may be modified according to a patient’s clinical course. Voriconazole should be avoided when feasible because of the potential for neurotoxicity.76
FAQ 5: What antiviral prophylaxis and monitoring strategies should be used for HBV, HCV, and HIV after CD19-targeted CAR–T-cell therapy?
Patients who are HBsAg+ or have detectable HBV DNA in blood should receive treatment with entecavir 0.5 mg orally daily starting pre CAR–T-cell infusion and for at least 6 months given that HBV can reactivate and cause severe disease in patients treated with B-cell–targeted drugs.24-26 The duration of treatment will depend on the patient’s clinical course. In patients who are anti-HBc+ but HBsAg and HBV DNA−, monitoring with serum nucleic acid testing for HBV DNA and alanine aminotransferase every 1 to 3 months can be considered as an alternative to entecavir prophylaxis. Patients in the aforementioned category who are also anti-HBs+ likely have even lower reactivation risk.26 Patients with chronic HCV should be referred to a specialist to discuss HCV monitoring and timing of anti-HCV therapy; a full discussion is beyond the scope of this review. There are 2 published reports of outcomes after CD19-targeted CAR–T-cell therapy in 3 individuals with HBV infection and 1 individual with HCV infection.25,27 All patients were successfully managed without serious complications except for 1 patient who died secondary to HBV reactivation after stopping prophylaxis 1 month post CAR–T-cell infusion. There are reports of successful CD19-targeted CAR–T-cell therapy in HIV-infected patients with large B-cell lymphoma, but the published experience remains limited.28,29 Treatment and monitoring of patients with HIV should be performed in consultation with an infectious disease (ID)/HIV expert prior to and after CAR–T-cell therapy.
FAQ 6: What antifungal prophylaxis should be used after CD19-targeted CAR–T-cell therapy?
We recommend antifungal prophylaxis with fluconazole 200 mg orally daily during periods of severe neutropenia (ANC <0.5 × 109/L; Figure 2) until neutrophil recovery. Micafungin 50 mg IV daily can be used as an alternative for patients with contraindications to fluconazole therapy. A mold-active azole may be required under certain circumstances, such as for patients with severe neutropenia for >3 weeks or who require additional immunosuppression after CD19-targeted CAR–T-cell therapy (see FAQ 13 and Table 2).
Patients already taking trimethoprim-sulfamethoxazole for Pneumocystis jirovecii pneumonia prophylaxis may continue through periods of neutropenia after CAR–T-cell therapy. If there is concern for potential myelosuppression, trimethoprim-sulfamethoxazole should be started after patients achieve an ANC >0.5 × 109/L and continued for at least 6 months (Figure 2). A suggested regimen is 1 double-strength tablet orally 3 times weekly. Prophylaxis for P jirovecii pneumonia should be started in all patients by day 28 after CAR–T-cell infusion. Alternative agents for patients with a sulfa allergy or prolonged cytopenias include aerosolized pentamidine 300 mg once monthly, dapsone 100 mg orally daily, and atovaquone 1500 mg orally daily.
FAQ 7: How should immunoglobulin levels be monitored and replaced in patients receiving CD19-targeted CAR–T-cell therapy?
The utility of prophylactic IgG infusions after CD19-targeted CAR–T-cell therapy is unknown, and there remains equipoise in the approach to IgG monitoring and replacement strategies. The direct effect of CD19-targeted CAR–T-cell therapy on total and pathogen-specific IgG production is incompletely understood. Long-lived plasma cells that produce the majority of antibodies to previously encountered pathogens may not be affected by CD19-targeted CAR–T-cell therapy due to low surface expression of CD19 (Figure 1).30-34 However, children with fewer established plasma cell clones might be more susceptible to infections after CD19-targeted CAR–T-cell therapy than adults.35,36 Prophylactic IgG has regulatory approval in select immunocompromised populations based on studies demonstrating reduced rates of serious bacterial infections but is not routinely recommended in cancer patients or HCT recipients.37-42 Thus, use of prophylactic IgG must be balanced with associated side effects, costs, and logistical challenges. We provide an algorithm to approach the management of hypogammaglobulinemia prior to and after CD19-targeted CAR–T-cell therapy in Figure 3 based in part on data from other contexts.39,43-49
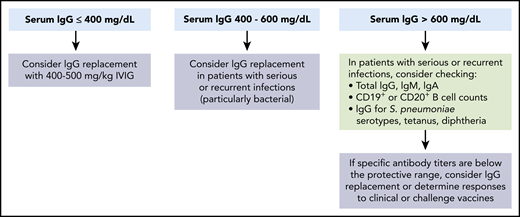
Indications for immunoglobulin replacement immediately prior to and for the first 3 months after CD19-targeted CAR–T-cell therapy. We suggest consideration of prophylactic immunoglobulin treatment prior to and after CD19-targeted CAR–T-cell therapy in patients with severe hypogammaglobulinemia (serum IgG <400 mg/dL). Higher thresholds can be considered in patients with serious or recurrent infections. Beyond the first 3 months after CD19-targeted CAR–T-cell infusion, we recommend consideration of prophylactic immunoglobulin treatment in patients with IgG ≤400 mg/dL and serious, persistent, or recurrent bacterial infections. Additionally, continuation of immunoglobulins could be considered in patients with IgG ≤400 mg/dL and persistent B-cell aplasia (≤20 cells per mm3 of CD19+ or CD20+ normal B cells in peripheral blood leukocytes). Reproduced from Hill et al49 with permission from Elsevier.
Indications for immunoglobulin replacement immediately prior to and for the first 3 months after CD19-targeted CAR–T-cell therapy. We suggest consideration of prophylactic immunoglobulin treatment prior to and after CD19-targeted CAR–T-cell therapy in patients with severe hypogammaglobulinemia (serum IgG <400 mg/dL). Higher thresholds can be considered in patients with serious or recurrent infections. Beyond the first 3 months after CD19-targeted CAR–T-cell infusion, we recommend consideration of prophylactic immunoglobulin treatment in patients with IgG ≤400 mg/dL and serious, persistent, or recurrent bacterial infections. Additionally, continuation of immunoglobulins could be considered in patients with IgG ≤400 mg/dL and persistent B-cell aplasia (≤20 cells per mm3 of CD19+ or CD20+ normal B cells in peripheral blood leukocytes). Reproduced from Hill et al49 with permission from Elsevier.
FAQ 8: What types of infections do patients get after CD19-targeted CAR–T-cell therapy?
Data regarding infectious complications after CD19-targeted CAR–T-cell therapy are limited, particularly beyond the first few months of treatment and in pediatric populations. We provide an overview of infection risk over time in Figure 4. Pivotal trials using tisagenlecleucel and axicabtagene ciloleucel report any infection in up to 55% of patients within the first 1 to 2 years, and infections of at least grade 3 severity in up to 33% of patients.1-6 Fatal infections are rare and reported in <5% of treated patients. In 3 studies focusing on infections after CD19-CAR–T-cell therapy using noncommercial products in children and/or adults with ALL, non-Hodgkin lymphoma, and/or chronic lymphocytic leukemia, the cumulative incidence of any microbiologically documented infection within the first month was as high as 40%.7,50,51 Most infections were bacterial, and almost all instances of bacteremia occurred within the first 2 weeks after CAR–T-cell infusion. Respiratory viral infections were the next most frequent. Cases of HSV and VZV reactivation in patients not adhering to acyclovir prophylaxis have been reported, but infections with other herpesviruses and double-stranded DNA viruses (eg, adenovirus, BK polyomavirus) appear to be infrequent. Invasive fungal infections occur in up to 8% of patients, including mold infections.52 Infection rates after the first month were less frequent and usually due to respiratory viruses. Reactivation of HBV with fulminant liver failure has been described, but the published cases of patients with chronic hepatitis or HIV infection, albeit limited, suggest the safety of CD19-targeted CAR–T-cell therapy in this context with appropriate management.25,27-29
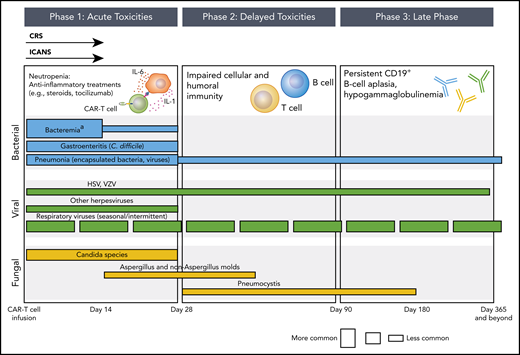
Phases of opportunistic infections in CD19-targeted CAR–T-cell therapy recipients.aApproximately 50% of bacteremia episodes are due to gram-positive organisms and 50% are due to gram-negative organisms. The conceptual model for this figure was adapted from Tomblyn et al.35
Phases of opportunistic infections in CD19-targeted CAR–T-cell therapy recipients.aApproximately 50% of bacteremia episodes are due to gram-positive organisms and 50% are due to gram-negative organisms. The conceptual model for this figure was adapted from Tomblyn et al.35
FAQ 9: What are the risk factors for infections after CD19-targeted CAR–T-cell therapy?
The most notable risk factor for infections after CD19-targerted CAR–T-cell therapy is the development of CRS, which is often accompanied by contributing factors such as additional immunosuppression and invasive procedures to manage critically ill patients.7,50,51 CRS-induced endothelial damage might initiate or facilitate some infections.53,54 Additional risk factors include a higher number of prior antitumor regimens. The contribution of persistent CD19+ B-cell depletion and hypogammaglobulinemia to infection risk is not well described in this population, and studies are challenging due to variable practices in immunoglobulin replacement.55 It is unclear whether infection risk differs between the commercially approved CD19-targeting products; the incidence and severity of CRS and ICANs, and the duration of B-cell aplasia, are the key factors that may affect acute and long-term infection risk.
FAQ 10: What are the infection risks associated with pharmacologic management of CRS and ICANS?
CD19 CAR–T-cell recipients may develop severe CRS or ICANS requiring high doses of corticosteroids and/or monoclonal antibodies targeting inflammatory cytokines (or their receptors) such as tocilizumab (IL-6 receptor antagonist), siltuximab (IL-6 antagonist), and anakinra (IL-1 receptor antagonist). Most experience with these monoclonal antibodies is in patients with autoimmune conditions receiving recurrent dosing, and studies have demonstrated a range of associated infections including M tuberculosis and invasive fungal infections.23,56 The direct contribution of anti-inflammatory monoclonal antibodies to immediate or delayed infections in the setting of limited dosing for treatment of CRS or ICANS is unknown.
FAQ 11: Can infection be distinguished from CRS?
Data exploring whether specific biomarkers can distinguish between CRS and infection, which have overlapping clinical presentations, are limited. One study found no differences in serum cytokine levels among subjects stratified by CRS grade and presence of infection.52 Luo et al found that a secondary increase in IL-6 levels in patients with CRS was associated with severe infection, and the authors proposed a prediction model of 3 cytokines (IL-8, IL-1β, and interferon-γ) to facilitate identification of infections after CAR–T-cell therapy.57 However, the study was small, and more work is needed to understand how to differentiate between CRS and infection.
FAQ 12: How should patients with symptoms of CRS and/or ICANS be managed for possible concurrent or new infections?
The majority of patients with CRS and/or ICANS will have concurrent neutropenia and present with symptoms indistinguishable from infection. Additionally, treatment of CRS and/or ICANS may mask typical signs and symptoms of infection. Thus, initial management needs to account for the possibility of infection with the understanding that CRS and/or ICANS will be the underlying cause in most instances. In general, blood cultures should be obtained along with other relevant diagnostic tests to evaluate for infections. Empiric broad-spectrum antibiotics should be initiated according to fever and neutropenia guidelines (Table 2).58 Escalation in therapy and diagnostic evaluation should be considered in all patients with progressive or new symptoms, particularly given that CRS is an important risk factor for subsequent infections after CD19-targeted CAR–T-cell therapy.7,14 Because the majority of patients will not have concomitant infections requiring broad-spectrum antimicrobial agents, de-escalation of empiric therapies should be addressed with an ID consultant on a daily basis.
We consider patients to be at particularly high risk for infections if they require >1 dose of tocilizumab, >3 days of high-dose corticosteroids (≥10 mg dexamethasone per day) within 7 days, methylprednisolone ≥1 g IV, or second-line agents such as anakinra (Table 2). We suggest ID consultation in all high-risk patients given the increased risk for bacteremia and opportunistic infections such as invasive molds and reactivation of latent infections such as CMV.7,14,52 In these individuals, we consider mold-active prophylaxis and weekly CMV monitoring for up to 1 month after immunosuppressive therapies are discontinued (Table 2).
Vaccination after CD19-targeted CAR–T-cell therapy
Clinical case continued
Our patient was discharged from the hospital on day 21, and evaluation on day 29 indicated that she was in a complete remission. Her serum total IgG concentration was 320 mg/dL, and she received another dose of IVIG. She was discharged home for management by her local oncologist. At the time of discharge, she was taking valacyclovir, trimethoprim-sulfamethoxazole, and posaconazole for antimicrobial prophylaxis. Over the next 6 months, she received 4 doses of IVIG for persistent hypogammaglobulinemia and recurrent upper respiratory tract infections without a microbiologic diagnosis. Her local oncologist called our immunotherapy long-term follow-up clinic to inquire about the need for vaccinations such as the 13-valent pneumococcal conjugate vaccine (Prevnar 13) and the recombinant zoster vaccine (Shingrix). She remained in complete remission and had not received any further antitumor therapies.
FAQs
FAQ 13: Should patients who received CD19-targeted CAR–T-cell therapy be (re)vaccinated?
The immunogenicity and safety of vaccinations after CD19-targeted CAR–T-cell therapy are unknown, and there are no data to guide vaccination approaches in this specific context. Patients treated with CD19-targeted CAR–T-cell therapy may have lower vaccine responses compared with healthy individuals, but vaccination may nonetheless prevent infections, decrease their severity, avoid hospitalizations, mitigate the need for prophylactic IgG therapy, and save lives. As such, the authors think that vaccination strategies after CD19-targeted CAR–T-cell therapy should be a critical component of a patient’s long-term care plan. This is particularly important given anticipated deficits in immunity to vaccine-preventable infections, such as prior HCT or in children who have not completed primary vaccine series.59-62 Additionally, prolonged CD19+ B-cell aplasia in this patient population may increase risk for infections with encapsulated bacteria, such as Haemophilus influenzae type b, Neisseria meningitidis, and Streptococcus pneumoniae. Whether CD19-targeted CAR–T-cell therapy directly impacts previously established humoral immunity to vaccine-preventable infections is unclear, although emerging data suggest that pathogen-specific antibody levels may not be impacted due to persistence of long-lived CD19− plasma cells.33,34,55
FAQ 14: What vaccinations should be administered before CD19-targeted CAR–T-cell therapy?
We recommend reviewing a patient’s vaccination history, including whether they are up to date with influenza and pneumococcal vaccinations, as part of the pre CAR–T-cell therapy evaluation.59 During the influenza season, we suggest administering the annual influenza vaccine at least 2 weeks prior to lymphodepleting chemotherapy. We do not recommend additional vaccines prior to CAR–T-cell therapy given the lower immunologic response anticipated in the context of patients with R/R malignancies receiving antitumor therapies.
FAQ 15: When should vaccinations be administered after CD19-targeted CAR–T-cell therapy?
The timing of vaccination after CAR–T-cell infusion will depend in part on a patient’s clinical course. In general, for patients who are in remission and do not require additional chemotherapy or HCT, vaccinations should be considered ≥6 months after CAR–T-cell infusion for killed/inactivated vaccines and ≥1 year for live and nonlive adjuvant vaccines. This recommendation is based on studies of vaccine responses in patients treated with rituximab (anti-CD20 monoclonal antibody),63-65 guidelines for vaccination of immunocompromised hosts including HCT recipients,59-62 and the kinetics of immune reconstitution after CD19-targeted CAR–T-cell therapy (see sFAQ 2).2,5,7,66 Patients who previously received an HCT should also meet established post-HCT criteria before starting vaccinations.35,59-61 Criteria to consider before initiating vaccinations are in Table 3.
Eligibility criteria for vaccinations after CD19-targeted CAR–T-cell therapy
| Criteria for vaccinations |
|---|
| Indications* |
| Killed/inactivated vaccines† |
| • ≥6 mo post CD19-targeted CAR–T-cell therapy |
| • ≥2 mo since last immunoglobulin treatment; a trial off of supplemental immunoglobulins can be considered in patients without chronic or serious bacterial infections in the preceding 6 mo |
| Live and nonlive adjuvant vaccines |
| • ≥1 y post CD19-targeted CAR–T-cell therapy |
| Contraindications* |
| Killed/inactivated vaccines† |
| • Supplemental immunoglobulins within the past 2 mo |
| • Receiving immunosuppressive therapy that reduces T-cell or B-cell function or active symptoms of graft-versus-host disease that requires treatment |
| • Administration of an anti-CD20 or anti-CD19 agent within the past 6 mo |
| • Actively receiving chemotherapy‡ |
| Live and nonlive adjuvant vaccines |
| • Administration of an anti-CD20 or anti-CD19 agent within the past 6 mo |
| • ≤1 y post CD19-targeted CAR–T-cell therapy |
| • ≤2 y post autologous or allogeneic HCT |
| • ≤1 y off all systemic immunosuppressive therapy |
| • ≤8 mo after last dose of supplemental immunoglobulins |
| • Absolute CD4+ T-cell count ≤200 cells/mm3 |
| • Absolute CD19+ or CD20+ B-cell count ≤20 cells/mm3 |
| • Actively receiving chemotherapy‡ |
| Criteria for vaccinations |
|---|
| Indications* |
| Killed/inactivated vaccines† |
| • ≥6 mo post CD19-targeted CAR–T-cell therapy |
| • ≥2 mo since last immunoglobulin treatment; a trial off of supplemental immunoglobulins can be considered in patients without chronic or serious bacterial infections in the preceding 6 mo |
| Live and nonlive adjuvant vaccines |
| • ≥1 y post CD19-targeted CAR–T-cell therapy |
| Contraindications* |
| Killed/inactivated vaccines† |
| • Supplemental immunoglobulins within the past 2 mo |
| • Receiving immunosuppressive therapy that reduces T-cell or B-cell function or active symptoms of graft-versus-host disease that requires treatment |
| • Administration of an anti-CD20 or anti-CD19 agent within the past 6 mo |
| • Actively receiving chemotherapy‡ |
| Live and nonlive adjuvant vaccines |
| • Administration of an anti-CD20 or anti-CD19 agent within the past 6 mo |
| • ≤1 y post CD19-targeted CAR–T-cell therapy |
| • ≤2 y post autologous or allogeneic HCT |
| • ≤1 y off all systemic immunosuppressive therapy |
| • ≤8 mo after last dose of supplemental immunoglobulins |
| • Absolute CD4+ T-cell count ≤200 cells/mm3 |
| • Absolute CD19+ or CD20+ B-cell count ≤20 cells/mm3 |
| • Actively receiving chemotherapy‡ |
Patients who previously received an HCT should also meet HCT-related criteria. These recommendations are irrespective of ongoing CD19+ B-cell aplasia.
For seasonal influenza vaccination, all patients should receive the annual vaccine irrespective of the above criteria.
Vaccination may be considered in patients who are receiving certain therapies that do not suppress T-cell and B-cell responses, such as checkpoint inhibitors, immunomodulatory agents (eg, lenalidomide), tyrosine kinase inhibitors, and select other agents.
FAQ 16: What vaccines should be administered after CD19-targeted CAR–T-cell therapy?
Based on the immunologic deficits after CD19-targeted CAR–T-cell therapy and vaccination guidelines in cancer patients (see FAQ 15), we recommend vaccinating all patients who received CD19-targeted CAR–T-cell therapy for S pneumoniae; hepatitis A virus and HBV (if susceptible); and Clostridium tetani, Corynebacterium diphtheriae, and Bordetella pertussis. For patients seropositive for VZV or who have a history of chickenpox or shingles, vaccination with Shingrix in adults ≥50 years old can be considered. Administration of other indicated vaccines should be considered, especially in children and prior HCT recipients. Conjugated vaccines should be preferentially used when available given higher response rates in immunocompromised patients compared with polysaccharide vaccines.67,68
FAQ 17: How should patients be vaccinated if they previously received an HCT or did not complete their primary vaccine series?
Vaccination strategies after CD19-targeted CAR–T-cell therapy may differ depending on whether a patient previously received an HCT. In patients who never received an HCT and were previously vaccinated, or patients with a prior HCT who completed post-HCT vaccinations, one can consider an approach that allows for the possibility of preserved immunity and/or the ability to generate a boosted response with a single vaccination as depicted in Figure 5A. An approach for patients who received an HCT prior to CD19-targeted CAR–T-cell therapy and did not complete post-HCT vaccines is depicted in Figure 5B. Suggested vaccination schedules for children and adults are detailed in supplemental Tables 1 and 2. These strategies were recently implemented at Fred Hutchinson Cancer Research Center. Given the lack of data pertaining to vaccine responses after CD19-targeted CAR–T-cell therapy, checking serum antibody titers may guide future practice. In patients without evidence of vaccine responses (Figure 5 footnote b), additional vaccinations should be deferred pending objective evidence of immune reconstitution (Table 4).
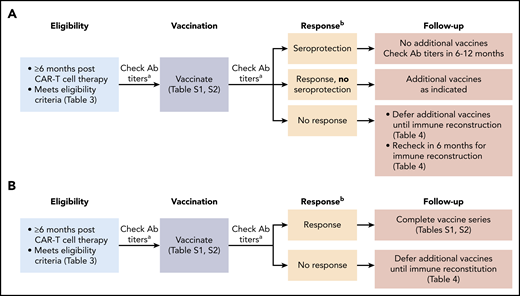
Possible approaches to vaccination strategies after CD19-targeted CAR–T-cell therapy. (A) Possible vaccination approach in CD19-targeted CAR–T-cell therapy recipients who have no history of prior HCT or who completed post-HCT vaccines. (B) Possible vaccination approach in CD19-targeted CAR–T-cell therapy recipients who have a history of prior HCT and did not complete post-HCT vaccines. aCheck serum IgG titers to S pneumoniae (23 serotypes), tetanus toxoid, hepatitis A virus, and HBVsAg. bA response is defined as achieving the following for all administered vaccines: for non–S pneumoniae vaccines: at least twofold increase in IgG from prevaccination to 1 to 2 months postvaccination or achieving a seroprotective IgG level at 1 to 2 months postvaccination77 ; for the S pneumoniae vaccine (Prevnar 13): at least twofold increase in IgG from prevaccination to 1 month postvaccination, achieving an IgG >1.3 µg/mL for ≥50% of the Prevnar 13 serotypes,77 or as defined by the testing laboratory. Ab, antibody. This figure was adapted from the Fred Hutchinson Cancer Research Center Standard Practice Manual.78
Possible approaches to vaccination strategies after CD19-targeted CAR–T-cell therapy. (A) Possible vaccination approach in CD19-targeted CAR–T-cell therapy recipients who have no history of prior HCT or who completed post-HCT vaccines. (B) Possible vaccination approach in CD19-targeted CAR–T-cell therapy recipients who have a history of prior HCT and did not complete post-HCT vaccines. aCheck serum IgG titers to S pneumoniae (23 serotypes), tetanus toxoid, hepatitis A virus, and HBVsAg. bA response is defined as achieving the following for all administered vaccines: for non–S pneumoniae vaccines: at least twofold increase in IgG from prevaccination to 1 to 2 months postvaccination or achieving a seroprotective IgG level at 1 to 2 months postvaccination77 ; for the S pneumoniae vaccine (Prevnar 13): at least twofold increase in IgG from prevaccination to 1 month postvaccination, achieving an IgG >1.3 µg/mL for ≥50% of the Prevnar 13 serotypes,77 or as defined by the testing laboratory. Ab, antibody. This figure was adapted from the Fred Hutchinson Cancer Research Center Standard Practice Manual.78
Immune reconstitution criteria for considering repeat vaccination attempts in nonresponders
| Criteria |
|---|
| Detectable serum IgA* (>6 mg/dL) AND |
| CD19+ or CD20+ B-cell count >20 cells/mm3AND |
| CD4+ T-cell count >200 cells/mm3 |
| Criteria |
|---|
| Detectable serum IgA* (>6 mg/dL) AND |
| CD19+ or CD20+ B-cell count >20 cells/mm3AND |
| CD4+ T-cell count >200 cells/mm3 |
Indicates potential ability to “class switch.”
Considerations for CAR–T-cell therapies for other diseases
The success of CD19-targeted CAR–T-cell therapies has fueled extensive investigations into CAR-T cells targeting other hematologic and solid malignancies.34 Most relevant to this manuscript are products targeting B-cell maturation antigen (BCMA) for R/R multiple myeloma.69-71 In contrast to CD19, which is present on early lineage B cells but less so on plasma cells,31 BCMA is selectively expressed by plasma cells (Figure 1B).72 Thus, BCMA-targeted CAR-T cells may have a greater impact on preexisting pathogen-specific immunity while preserving naïve and memory B-cell populations.
Conclusions
As CAR–T-cell targets and chemotherapeutic approaches (eg, combination with checkpoint inhibitors) advance, the field must be diligent with regard to the evolving landscape of infection risk and refine preventive strategies accordingly. Decades of pioneering work in the field of HCT, one of the first “immunotherapies,” has taught us that preventing infectious complications is critical to improving long-term outcomes.73 Although much work remains, we hope that the enumerated strategies will provide a starting point for achieving this goal.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Paul Carpenter, Jae Park, Janet Englund, David Maloney, Cameron Turtle, Merav Bar, Leona Holmberg, Steven Pergam, Kevin Curran, Tobias Hohl, and Michael Boeckh for their input, as well as Ashley Sherrid for assistance with figures.
This work was supported in part by the following grants from the National Institutes of Health: National Institute of Allergy and Infectious Diseases grant K23AI119133 and National Cancer Institute grant U01CA247548 (J.A.H.), and National Cancer Institute Cancer Center support grants P30CA0087-48 and P30CA015704-44.
Authorship
Contribution: J.A.H. conceived the FAQ format and wrote the manuscript; and S.K.S. critically read and edited the manuscript and provided additional in-depth expertise on infection-prevention strategies.
Conflict-of-interest disclosure: J.A.H. has served as a consultant for Nohla Therapeutics, Inc, Amplyx, and Gilead Sciences, and has received research support from Nohla Therapeutics, Karius, and Takeda (formerly Shire). S.K.S. declares no competing financial interests.
Correspondence: Joshua A. Hill, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Mail Stop E-400, Seattle, WA 98109; e-mail: jahill3@fredhutch.org.
