

In this issue of Blood, identify a set of 22 genes, including negative regulators of T-cell receptor (TCR) signaling, downregulated with promoter hypermethylation in human T-cell lymphotropic virus-1 (HTLV-1) infected CD4+ T cells. The authors then show that the use of hypomethylating agents (HMAs) inhibits adult T-cell leukemia-lymphoma (ATL) growth in xenograft mice.1
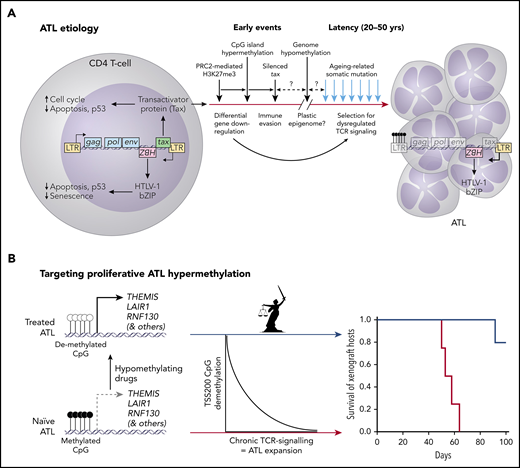
(A) The etiology of HTLV-1 infection (splicing and scale are ignored in the simplified virus genome) and conversion to ATL. (B) ATL etiology involves promoter CpG hypermethylation that silences downregulators of TCR signaling, including Themis, the Titaness personification of natural law. Expansion of chronic ATL xenografts in mice was significantly inhibited by HMA therapy with OR-21.
(A) The etiology of HTLV-1 infection (splicing and scale are ignored in the simplified virus genome) and conversion to ATL. (B) ATL etiology involves promoter CpG hypermethylation that silences downregulators of TCR signaling, including Themis, the Titaness personification of natural law. Expansion of chronic ATL xenografts in mice was significantly inhibited by HMA therapy with OR-21.
ATL is a highly malignant neoplasm that originates from CD4+ T cells infected by HTLV-1, 20 to 50 years postinfection. HTLV-1 is endemic in parts of Japan, the Caribbean, Central and South America, Central Africa, Romania, Iran, and Papua New Guinea. HTLV-1 confers a 3% to 5% lifetime risk for ATL in the estimated 10 to 20 million people carrying the virus.2 HTLV-1 randomly inserts as a retroviral provirus into the genome of infected cells. In addition to canonical gag, pol, and env genes, HTLV-1 encodes several accessory genes; 2 of these, tax and HBZ (see figure panel A), are oncogenic in transgenic mice. The complex Tax protein interactome, which includes kinases and E3 ligases, activates NF-κB, cyclin-dependant kinases, autophagy, and anti-apoptosis factors to enhance host cell proliferation. The HBZ protein modulates Tax activities and inhibits senescence and apoptosis.3 Early in infection, Tax orchestrates widespread PRC2-mediated histone odification (ie, H3K27me3), downregulating targeted promoters.4 This probably precedes CpG methylation changes that are the focus of Watanabe et al. Chromatin remodeling eventually affects the HTLV-1 5′ LTR, inducing viral latency and silencing tax, but not transcription of HBZ from the 3′ LTR (see figure panel A). The self-reinforcing epigenome thus established produces a stably disturbed transcriptome diabolically similar to that induced by Tax itself and seems to ensure long-term host cell persistence in Tax’s absence. This phenomenon might have been selected by Tax’s high immunogenicity.4 The discovery of an aging-related somatic mutation signature in ATL suggested that neoplasia develops over decades, long after HTLV-1 latency, selecting for mutations (both Tax and HBZ inhibit p533 ) plus an epigenome that de-represses TCR signaling to promote ATL expansion5 (see figure panel A).
Diversity in clinical features and prognosis led to subclassification of ATL into smoldering, chronic and acute leukemic forms, and ATL lymphoma.6 Although smoldering and chronic ATL are relatively indolent (4-year overall survival rates, 52% and 36%), about one-half progress to aggressive ATL (acute and lymphoma; 4-year overall survival rates, 11% and 16%).7 Therapies include zidovudine plus interferon-alfa, monoclonal anti-CCR4 therapy, multiagent chemotherapy, or allogeneic hematopoietic stem cell transplantation, but cure is rarely achieved.8
Watanabe et al used Infinium BeadArrays to interrogate CpG methylation in HTLV-1 infected and uninfected CD4 T cells sorted by CADM1 and CD7 expression from healthy controls (n = 9), asymptomatic carriers (n = 3), and smoldering (n = 3), chronic (n = 4), and acute (n = 3) ATL patients. HTLV-1-infected cells progress from CADM1–/CD7+ (healthy T) through a CADM1+/CD7dim population to a CADM1+/CD7– population that increases from indolent to acute ATL. HTLV-1-infected T cells were distinguished by CpG hypomethylation profiles using 20 000 CpG probes randomly sampled from the total of 470 870. Although global hypomethylation characterized infected cells, there was no significant difference in global hypomethylation levels between clinical groups, so its biological significance as well as its etiology remain unclear. Delving deeper, 12 025 hypermethylated and 33 581 hypomethylated regions were identified in infected vs healthy T cells that were shared in all ATL patients; aggressive and indolent ATL clustered using the hypermethylated but not hypomethylated regions. These included 1207 hypermethylated CpG islands lying within 200 bp 5′ of transcription start sites (“TSS200”), with increasing hypermethylation as ATL progressed. A set of 22 downregulated genes with corresponding TSS200 hypermethylation included negative regulators of TCR signaling: LAIR, RNF130, and THEMIS. Dysregulation of TCR signaling is an established ATL hallmark.5 Downstream effectors of TCR signaling were shown to be either persistent or constitutively active in ATL cell lines and amenable to downregulation by forced expression of THEMIS.
These data motivated experiments to reverse hypermethylation using clinically available hypomethylating agents: azacitidine (AZA) and decitabine (DAC), plus 2 new decitabine prodrugs, OR12 and OR21, which have the potential for oral bioavailability.9 Encouraged by their in vitro data, Watanabe et al inoculated MT-2 ATL cells into mice subcutaneously and administered AZA, DAC, or OR21 intraperitoneally. They observed tumor reduction by all three HMAs, albeit with variable hematopoietic toxicity (least to most; AZA, OR21, DAC) and hypomethylation of THEMIS TSS200 (least to most; AZA, OR21, DAC) at the doses used. With a view to potential long-term oral therapy, OR-21 was used (intraperitoneally, twice weekly for 100 days) to treat mice bearing peritoneal hCD45+/CADM1+ xenografts established by inoculating peripheral blood mononuclear cells from a patient with chronic ATL. OR21 produced a striking improvement in morbidity and mortality (see figure panel B).
These discoveries were possible because the authors focused their analysis on shared differentially methylated regions in infected/uninfected cells in patients across the disease spectrum; the devil lies in finding relevant detail. Disregarding the small cohort size, it is encouraging that epigenetically downregulated genes converged on pathways already implicated by orthogonal means and demonstrably amenable to therapeutic manipulation. The degree to which hypomethylation or addiction to TCR signaling dictate drug efficacy remains debatable; AZA was equally efficacious in reducing tumor volume despite significantly lower effect on CpG methylation in the THEMIS promoter than DAC or OR-21. Parenteral formulations of AZA and DAC are in regular clinical use for higher risk myelodysplastic syndrome, chronic myelomonocytic leukemia and low-blast acute myeloid leukemia (AML) and are an alternative for newly diagnosed AML in the elderly who have comorbidities that preclude use of intensive induction chemotherapy. Because HMA therapy is continued for as long as patients derive benefit, oral formulations are preferable, and oral AZA (CC-486) and decitabine and cytidine deaminase inhibitor combinations (eg, ASTX727) have advanced through to phase 3 trials, with the former showing promise as maintenance therapy following AML remission.10 OR-21 was shown to be bioavailable following intraduodenal administration to macaques and is potentially another oral HMA.1,9 Despite the limitations of their xenograft model, Watanabe et al demonstrate that a slumbering Themis can be roused to restore her natural law. Their data are a compelling precursor for clinical evaluation of HMAs in ATL.
Conflict-of-interest disclosure: J.E.P. has served on advisory boards and received honoraria or clinical trial funding from Celgene/BMS and Abbvie. C.J.J. declares no competing financial interests.
