

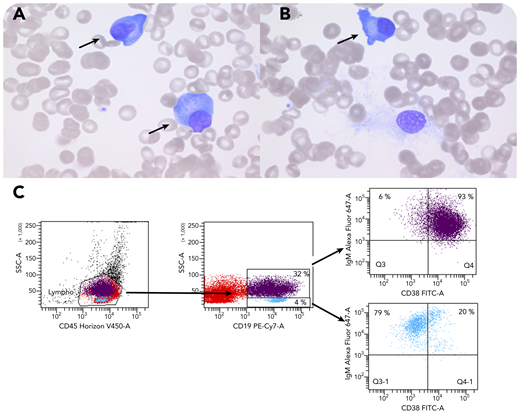
A 6-year-old boy with a history of B-cell non-Hodgkin lymphoma (Burkitt type) 2 years earlier presented with high fevers, hepatosplenomegaly, and trilineage cytopenia due to initial Epstein-Barr virus (EBV) infection. Elevated sCD25, ferritin, and triglycerides, together with hypofibrinogenemia, confirmed the diagnosis of EBV-triggered hemophagocytic lymphohistiocytosis (HLH), to which he succumbed. Bone marrow examination showed absent myelopoiesis and erythropoiesis with predominant T cells, hemophagocytosis, and immunohistochemically ∼25% CD38+CD20− EBV+ plasma cells. T and natural killer (NK) cells showed no SAP protein expression by flow cytometry, confirming the diagnosis of X-linked lymphoproliferative disease (XLP), genetically confirmed by a hemizygous variant (c.163C>T;p.Arg55*) in SH2D1A. This primary immunodeficiency is characterized by disturbed T:B-cell interaction, leading to impaired killing of EBV-infected B cells. It typically presents as EBV-triggered HLH, B-cell lymphoma, or hypogammaglobulinemia. The bone marrow smear (panels A-B; ×100 objective, original magnification ×1000; Wright-Giemsa stain) shows scattered plasmacytoid cells (panels A-B, arrows) in an otherwise aplastic bone marrow with hemophagocytosis as a typical finding in XLP with acute EBV infection. These cells represent EBV-infected CD19+CD38+IgM+ plasma blasts (panel C, purple), which are insufficiently killed by the SAP-deficient T and NK cells.
Primary immunodeficiencies can present with malignancy, and immunological and genetic workup at diagnosis may be indicated, as well as in male patients presenting with severe cytopenia and EBV infection.
A 6-year-old boy with a history of B-cell non-Hodgkin lymphoma (Burkitt type) 2 years earlier presented with high fevers, hepatosplenomegaly, and trilineage cytopenia due to initial Epstein-Barr virus (EBV) infection. Elevated sCD25, ferritin, and triglycerides, together with hypofibrinogenemia, confirmed the diagnosis of EBV-triggered hemophagocytic lymphohistiocytosis (HLH), to which he succumbed. Bone marrow examination showed absent myelopoiesis and erythropoiesis with predominant T cells, hemophagocytosis, and immunohistochemically ∼25% CD38+CD20− EBV+ plasma cells. T and natural killer (NK) cells showed no SAP protein expression by flow cytometry, confirming the diagnosis of X-linked lymphoproliferative disease (XLP), genetically confirmed by a hemizygous variant (c.163C>T;p.Arg55*) in SH2D1A. This primary immunodeficiency is characterized by disturbed T:B-cell interaction, leading to impaired killing of EBV-infected B cells. It typically presents as EBV-triggered HLH, B-cell lymphoma, or hypogammaglobulinemia. The bone marrow smear (panels A-B; ×100 objective, original magnification ×1000; Wright-Giemsa stain) shows scattered plasmacytoid cells (panels A-B, arrows) in an otherwise aplastic bone marrow with hemophagocytosis as a typical finding in XLP with acute EBV infection. These cells represent EBV-infected CD19+CD38+IgM+ plasma blasts (panel C, purple), which are insufficiently killed by the SAP-deficient T and NK cells.
Primary immunodeficiencies can present with malignancy, and immunological and genetic workup at diagnosis may be indicated, as well as in male patients presenting with severe cytopenia and EBV infection.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal