

Key Points
This large study of patients with verified CAD found a fourfold difference in prevalence and incidence between cold and warmer climate.
Rituximab-bendamustine therapy yielded 78% response rate, 53% complete response, response duration >88 months, and few late malignancies.

Abstract
We retrospectively studied 232 patients with cold agglutinin disease (CAD) at 24 centers in 5 countries. In Norway and a northern region of Italy, the study was close to being population-based. For the first time, we demonstrate fourfold differences between cold and warmer climates regarding prevalence (20 vs 5 cases/million) and incidence (1.9 vs 0.48 cases/million per year). Mean baseline hemoglobin level was 9.3 g/dL, but 27% had hemoglobin <8 g/dL. Identification of typical features of CAD-associated lymphoproliferative disorder in the bone marrow was greatly increased by centralized biopsy assessment. CAD seems to be associated with a slightly increased risk of venous thrombosis. This work includes a follow-up study of therapies, focusing on the long-term outcomes of the rituximab plus bendamustine and rituximab plus fludarabine regimens. Rituximab plus bendamustine therapy resulted in responses in 35 (78%) of 45 patients; 24 (53%) achieved complete response. Interestingly, these rates were still higher than observed in the original (2017) prospective trial, and we also found a shift toward deeper responses with time. This is explained by the prolonged time to response seen in many patients, probably related to long-lived plasma cells. In patients responding to rituximab-bendamustine, median response duration was not reached after 88 months, and estimated 5-year sustained remission was 77%. The regimen appeared safe regarding late-occurring malignancies. Rituximab plus fludarabine therapy seems to carry a higher risk of long-term adverse effects.
Introduction
The past 2 decades have seen great progress in the basic understanding and treatment of cold agglutinin disease (CAD).1-3 The autoantibodies in CAD are cold agglutinins (CAs), most often of the immunoglobulin Mκ (IgMκ) class, that are able to agglutinate erythrocytes at an optimum temperature of 3°C to 4°C.4-7 If the thermal amplitude of the CA autoantibody allows binding to its antigen in vivo, this will result in classical complement pathway-mediated autoimmune hemolytic anemia (AIHA) and, often, agglutination-mediated ischemic symptoms affecting the acral circulation.2,8-11 In most if not all patients, CA is monoclonal and produced by a very indolent, clonal B-cell lymphoproliferative disorder (LPD) of the bone marrow that usually displays the characteristics of “CAD-associated LPD.”12-14 According to recent definitions, the concept of CAD does not include secondary CA syndrome (CAS).1,2,15,16 Only CAD is studied in this work.
Despite previous studies, many questions remain unanswered regarding epidemiology, clinical and hematologic features, and outcomes.6,7,17,18 Although seasonal variations of hemolysis have been documented in individual cases,19-21 no direct evidence has been published for the notion that CAD is more prevalent or severe in colder climates. The term “cold” refers to the biologic properties of the autoantibody, not the clinical features.22,23 Previously published studies reported conflicting data on mortality and risk of thromboembolic events (TEs),6,17,18,24 although an increased risk of TEs has been well documented in warm-antibody AIHA.25-27 Good data on iron overload have not been provided.
Treatment is not always indicated, but historically, 70% to 80% of the patients have received therapy.1,2,6,7,28 Rituximab monotherapy has yielded response rates of ∼50%, only rare complete responses (CR) and a median response duration of 11 to 12 months.29,30 These results can be improved by adding bendamustine or fludarabine, but at a risk of short-term toxicity and with concerns about possible late adverse effects.31-33 Furthermore, these treatments are often rather slow-acting.31,32 Bortezomib has also shown a favorable effect, although response rates seem to be relatively low after monotherapy; 6 of 19 patients responded in a prospective trial.34 No study has been published on the effect of Bruton tyrosine kinase inhibitors and other novel specific B-cell targeting agents in CAD.
Complement-directed therapies are promising but still investigational.11,35 Blocking the terminal complement cascade by using the anti-C5 monoclonal antibody, eculizumab, produced a dramatic reduction in the need for transfusions and an increase in hemoglobin (Hb) levels that was modest, but higher compared with trials in paroxysmal nocturnal hemoglobinuria.24,36-38 Inhibition at the C1 or C3 level should be expected to work better,10 and favorable clinical results have recently been published with the anti-C1s monoclonal antibody, sutimlimab (BIVV009).35,39 A phase 2 clinical trial of the pegylated cyclic peptide, pegcetacoplan, an inhibitor of C3, has also shown promising results.40 These therapies cannot be expected to relieve the agglutination-mediated circulatory symptoms, which are not complement-dependent.11,41 Among supportive therapies, temporary use of erythropoietin has the potential to result in a meaningful increase in Hb levels, at least in some patients.42,43
We undertook a comprehensive, observational study to improve our knowledge of the epidemiologic, clinical, and laboratory findings in CAD. We also wanted to provide “real-world” information on the long-term outcomes of previously studied therapies, with focus on the rituximab-fludarabine and rituximab-bendamustine combinations.31,32
Methods
Study design
Between June 2017 and January 2019, we performed a retrospective, multinational, multicenter study of as many CAD patients who could be identified in Norway and Lombardy, a northern region of Italy, as well as known patients from 4 centers in the United Kingdom, Finland, and Denmark. We analyzed baseline and follow-up data from the first relevant examination until the time of last data collection or the patient’s death. Informed consent was obtained in all but deceased patients. The study protocol was approved by the Regional Committee for Medical and Health Research Ethics, South East Norway (approval #2016/618) and subsequently by the relevant ethics committees in the other countries. Patient-related data were deidentified, but a patient number was assigned and an identification key was retained at each center.
Identification of patients
All Norwegian hospitals with specialized hematology services participated in the study. In Norway, all hematology is taken care of by public hospitals run by governmental health care trusts, and every patient diagnosed with CAD will have been seen at 1 of these centers. Patients were also accrued at a large Italian university hospital with nearly population-based responsibility for AIHA care in most of the Lombardy region. The National Health Service of Italy is public and treatments for rare diseases are fully reimbursed. In Lombardy, the participating center is a recognized reference for the expertise acquired and receives patients in second opinion from the territory as well as follows directly most of those nearby.
Patients with a CAD diagnosis were found by contacting the hematologist(s) responsible for AIHA care at each center. Each local investigator performed an electronic search for patients with AIHA (International Classification of Diseases-10 diagnosis D59.1), and those diagnosed with CAD were then identified by reviewing the medical records. Patients who had been hospitalized as well as those seen only as outpatients were considered for the study.
Additional patients were accrued from 2 university hospital centers in London, United Kingdom; 1 university hospital in Finland; and 1 in Denmark. Based on this identification procedure, data from Norway and Italy were considered being as population-based as possible, whereas patient data from the United Kingdom, Denmark, and Finland were considered as possibly selected and were not used for epidemiologic calculations.
Inclusion and exclusion criteria
Patients who were alive by December 31, 2018, or had died between January 2007 and December 2018, were included if they met the specific diagnostic criteria defined in the study protocol. These criteria were chronic hemolysis, positive direct antiglobulin test (DAT), monospecific DAT positive for C3d, CA titer ≥64, and absence of any cause of secondary CAS (such as clinically or radiologically overt lymphoma, other active cancer, or recent infection with Mycoplasma pneumoniae or Epstein-Barr virus). Demonstration of a low-grade bone marrow LPD by histology or flow cytometry was not an exclusion criterion. Patients who fully met these criteria were classified as having “confirmed CAD.”
Some reported patients met all but 1 criterion (usually by failing the CA titer requirement) but were still perceived as having CAD, supported by additional features such as acrocyanosis, IgMκ gammopathy, and typical bone marrow findings. These were included as “probable CAD” provided consensus between 2 experts (S.B., W.B., S.D., and G.E.T.). Remaining patients who had been diagnosed with CAD by their hospitals but did not meet the predefined diagnostic criteria were classified as “not or probably not CAD” and were excluded.
Treatment outcomes
For therapy given within the rituximab plus fludarabine or rituximab plus bendamustine trials, we used previously published response definitions and graded responses into CR, partial response (PR), and no response.1,31,32 The CR criteria included complete histologic and flow cytometric regression of any detectable bone marrow LPD. For treatment administered outside these trials, response was defined as a stable (≥2 consecutive measurements) increase in Hb levels of ≥2.0 g/dL or to the normal range and no transfusion requirement. Responses were not graded in these cases.
Complete treatment outcomes definitions, sources of data, and statistics are described in the supplemental Data on the Blood Web site.
Results
Epidemiology
Table 1 shows prevalence and incidence. Of 251 patients reported to the study, 232 (92.5%) had CAD and were included; 207 (82.5%) had confirmed CAD; and 25 (10.0%) had probable CAD. Mean age was 67 years at disease onset, 68 at diagnosis, and 76 at the time of the study (supplemental Table 1). The male-to-female ratio was 0.56. Among 191 patients with complete data on disease duration, 115 (60.2%) had been diagnosed within 1 year of clinical onset; longest time to diagnosis was 32 years. Mean duration from onset of anemia or symptoms to death or final data collection was 10 years (range, 0-57). The mean follow-up time was 8 years from diagnosis (median, 6 years; range, 0-32).
Prevalence and incidence
| Population, 106 | Prevalence, cases/106 inhabitants | Incidence, cases/106 inhabitants/y | Outdoor temperature, °C, yearly-average | |
|---|---|---|---|---|
| Norway | 5.32 | 20.5 | 1.9 | 6.0* |
| Lombardy, Italy | 7.0† | 5.0 | 0.48 | 13.1 |
| Population, 106 | Prevalence, cases/106 inhabitants | Incidence, cases/106 inhabitants/y | Outdoor temperature, °C, yearly-average | |
|---|---|---|---|---|
| Norway | 5.32 | 20.5 | 1.9 | 6.0* |
| Lombardy, Italy | 7.0† | 5.0 | 0.48 | 13.1 |
Calculation of prevalence was based on the number of patients still alive at the end of the study period. The yearly number of newly diagnosed cases was approximately constant from 2007 and was used to estimate incidence.
Heterogeneous. Estimate based on yearly-average in Oslo, Bergen, and Trondheim.
Refers to the relevant part of the region.
Comorbidity
Comorbidities were reported in 156 patients (67.2%), including autoimmune diseases other than AIHA in 27 (11.6%), cured or inactive malignancies in 15 (6.5%), and hepatitis B or C in 6 (2.6%) (supplemental Data; supplemental Table 2).
Clinical features
Of 210 patients with relevant available data, cold-induced circulatory symptoms were recorded in 120 patients (51.7%) at or before diagnosis and 108 (46.6%) at final data collection. Details and grading are shown in supplemental Table 3. We classified the patients into 3 clinical phenotypes as shown in Table 2. These phenotypes did not show any association with the demonstration of a clonal LPD by bone marrow histology or the response to B-cell-directed therapies.
Clinical phenotypes
| Clinical phenotype | Frequency | ||
|---|---|---|---|
| Type | Definition | n | % |
| 1 | Hemolytic anemia with circulatory symptoms grade 1 or absent | 146 | 69.5 |
| 2 | Hemolytic anemia with circulatory symptoms grade 2-3 | 44 | 21.0 |
| 3 | Circulatory symptoms with compensated hemolysis | 20 | 9.5 |
| All patients with available data | 210 | 100.0 | |
| Clinical phenotype | Frequency | ||
|---|---|---|---|
| Type | Definition | n | % |
| 1 | Hemolytic anemia with circulatory symptoms grade 1 or absent | 146 | 69.5 |
| 2 | Hemolytic anemia with circulatory symptoms grade 2-3 | 44 | 21.0 |
| 3 | Circulatory symptoms with compensated hemolysis | 20 | 9.5 |
| All patients with available data | 210 | 100.0 | |
Circulatory symptoms grade 1, acrocyanosis only; grade 2, Raynaud-like symptoms interfering with daily living; grade 3, gangrene or ulcerations.
Laboratory findings
Table 3 shows Hb levels, markers of hemolysis, IgM concentrations, and CA titers at baseline. Mean Hb level was 9.3 g/dL (median, 9.2; range, 4.5-15.3). Sixty-two patients (26.7%) presented with Hb <8.0 g/dL and 89 patients (38.4%) with Hb ≥10 g/dL (supplemental Figure 1). DAT was positive for C3d in all cases by definition, weakly positive for IgG in 46 cases (19.8%), and positive for IgM in 12 cases (5.2%). Data on monoclonal immunoglobulins are provided in supplemental Table 4.
Hemoglobin and markers of disease activity at diagnosis
| Median (range) | Within reference range, % of patients | |
|---|---|---|
| Hemoglobin, g/dL | 9.2 (4.5-15.3) | 10.0 |
| Total bilirubin, μmol/L | 36 (5-136) | 14.3 |
| Lactate dehydrogenase, U/L | 380 (117-2026) | 9.7 |
| Haptoglobin, g/L | <0.1 (<0.1-9.0) | 11.2 |
| Reticulocytes, 109 cells/L | 147 (13-778) | 20.7 |
| Total IgM, g/L | 3.2 (0.2-74) | 34.6 |
| Cold agglutinin titer at 4°C | 512 (16-819,200) | 8.6 |
| Median (range) | Within reference range, % of patients | |
|---|---|---|
| Hemoglobin, g/dL | 9.2 (4.5-15.3) | 10.0 |
| Total bilirubin, μmol/L | 36 (5-136) | 14.3 |
| Lactate dehydrogenase, U/L | 380 (117-2026) | 9.7 |
| Haptoglobin, g/L | <0.1 (<0.1-9.0) | 11.2 |
| Reticulocytes, 109 cells/L | 147 (13-778) | 20.7 |
| Total IgM, g/L | 3.2 (0.2-74) | 34.6 |
| Cold agglutinin titer at 4°C | 512 (16-819,200) | 8.6 |
Flow cytometry in bone marrow aspirates showed a median ratio between κ and λ positive B-lymphocytes of 7.0 (range, 0.2-839) in 102 patients with sufficiently detailed data; the ratio was >3.5 in 84 patients (82%). Figure 1 shows bone marrow biopsy findings in 176 patients with available histology data. Assessment for MYD88 L265P mutation was reported in 16 patients only; this mutation was not detected in any of 14 patients with CAD-associated LPD but was found in 2 cases of lymphoplasmacytic lymphoma.
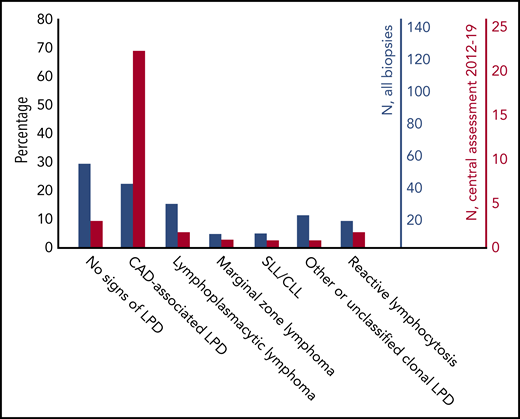
Bone marrow biopsy findings in 176 patients with available data. Comparison by percentage. Centralized revision increased the detection rate of clonal LPD and the percentage of biopsies interpreted as CAD-associated LPD. Blue bars, all biopsies. Red bars, biopsies that underwent a centralized review from 2012 through 2019. CLL, chronic lymphocytic leukemia; SLL, small lymphocytic B-cell lymphoma.
Bone marrow biopsy findings in 176 patients with available data. Comparison by percentage. Centralized revision increased the detection rate of clonal LPD and the percentage of biopsies interpreted as CAD-associated LPD. Blue bars, all biopsies. Red bars, biopsies that underwent a centralized review from 2012 through 2019. CLL, chronic lymphocytic leukemia; SLL, small lymphocytic B-cell lymphoma.
Serum levels of complement C3 and C4, C-reactive protein, folate, ferritin, and transferrin saturation are listed in supplemental Table 5. Serum folate levels were above the lower limit of normal in 97% of the patients, even in the absence of supplementation. Ferritin levels were higher in transfused than in nontransfused patients (median, 524 μg/L vs 219 μg/L; P < .001). Confirmed organ complications of iron overload were not reported.
Survival and complications
Seventy-two patients (31%) died during follow-up, whereas 160 patients (69%) were still alive at the end of the observation period. In 8 patients (11% of the deceased, 3.5% of total), death was considered caused by CAD (hemolytic anemia or underlying low-grade LPD) or its complications (transformation and treatment complications included). The survival curve for all patients is shown in Figure 2; median estimated survival was 16 years from diagnosis (range, 0-32), and the 5-year survival was estimated to 83%.
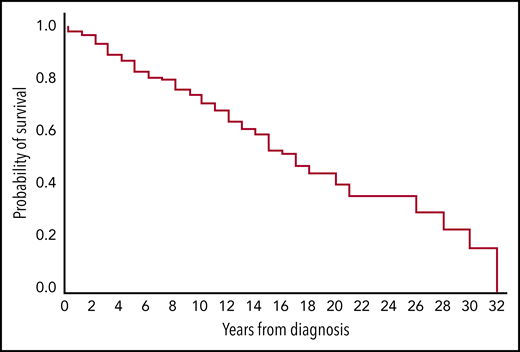
Estimated survival from diagnosis in 232 patients with cold agglutinin disease. Kaplan-Meier plot. Censored observations (n = 161) not marked.
Estimated survival from diagnosis in 232 patients with cold agglutinin disease. Kaplan-Meier plot. Censored observations (n = 161) not marked.
During follow-up, development of diffuse large B-cell lymphoma (DLBCL) occurred in 8 patients (3.4%). One patient (0.4%) was diagnosed with multiple myeloma, which was shown to be unrelated to the CAD-associated B-cell clone.44 Two patients (0.9%) developed acute myelogenous leukemia (AML) and 1 (0.4%) acute lymphoblastic leukemia; all 3 died of leukemia. Development of myelodysplastic syndrome (MDS) was recorded in 2 patients (0.9%); 1 of these died of MDS and 1 transformed to AML. On reexamination of data, MDS was found to have started before CAD in the latter patient. We observed 18 cases of nonhematologic malignancies (7.7%) after CAD diagnosis: 7 basal or squamous cell skin cancers, 3 cases of colon carcinoma, 2 cases of renal carcinoma, and 6 single occurrences of other cancers.
Thirty patients (12.9%) had experienced a total of 38 TEs during follow-up: 14 venous (VTE) (37% of TEs) and 24 (63%) arterial (ATE). Eight VTEs (57%) were complicated by pulmonary embolism. VTEs in atypical locations (such as splanchnic or cerebral) were not observed. Among the ATEs, 11 individuals experienced a total of 12 strokes (50% of ATEs), 10 had a myocardial infarction (MI) (42% of ATEs), and 2 had a peripheral embolic event. Occurrence of TE was not associated with severity of anemia (Hb <8 g/dL vs Hb ≥8 g/dL).
Therapy outcomes
Fifty-six patients (24.1% of 232) had not been treated, whereas 175 (75.9%) had received 1 to 6 (median, 2) lines of therapy (supplemental Figure 2; supplemental Table 5). Of 104 patients who had received 1 to 4 (median, 1) lines of rituximab monotherapy, 61 (59%) had responded at least once. Among 51 patients who had received this treatment more than once, 3 (6%) had never responded, 28 (55%) had achieved 1 response and thereafter been unresponsive, whereas 20 (39%) had experienced repeated responses after 2 to 4 courses. The median response duration was 15 months (range, 1-60).
All 29 patients who participated in the Norwegian 2010 rituximab-fludarabine trial32 were also included in this study. On reevaluation of outcomes, 18 patients (62%) had responded: 11 (38%) had achieved CR and 7 (24%) PR. Estimated response duration is displayed in Figure 3. Median estimated response duration was 77 months (range, 18-180), and estimated 5-year sustained remission was 71%. Frequencies of late-occurring malignancies appear from Table 4.

Probability of sustained remission in patients who have responded to 4 cycles of rituximab plus fludarabine. Kaplan-Meier plot showing a median estimated response duration of 77 months (range, 18-180) and a 5-year estimated sustained response rate of 71%.
Probability of sustained remission in patients who have responded to 4 cycles of rituximab plus fludarabine. Kaplan-Meier plot showing a median estimated response duration of 77 months (range, 18-180) and a 5-year estimated sustained response rate of 71%.
Late-onset malignancies according to treatment groups
| Treatment | Type of late-occurring malignancy; patients, n (%) | |||||
|---|---|---|---|---|---|---|
| Group | Patients, n | DLBCL | AML/ALL | MDS | Solid tumors | All malignancies |
| Rituximab plus fludarabine (follow-up of prospective trial) | 29 | 2 (7) | 2 (7) | 1 (3.5) | 4 (14) | 9 (31) |
| Rituximab plus bendamustine (follow-up of prospective trial) | 45 | 2 (4.5) | 0 (0) | 0 (0) | 2 (4.5) | 4 (9) |
| All patients | 232 | 8 (3.5) | 2 (1) | 2 (1) | 18 (8) | 29 (13) |
| Treatment | Type of late-occurring malignancy; patients, n (%) | |||||
|---|---|---|---|---|---|---|
| Group | Patients, n | DLBCL | AML/ALL | MDS | Solid tumors | All malignancies |
| Rituximab plus fludarabine (follow-up of prospective trial) | 29 | 2 (7) | 2 (7) | 1 (3.5) | 4 (14) | 9 (31) |
| Rituximab plus bendamustine (follow-up of prospective trial) | 45 | 2 (4.5) | 0 (0) | 0 (0) | 2 (4.5) | 4 (9) |
| All patients | 232 | 8 (3.5) | 2 (1) | 2 (1) | 18 (8) | 29 (13) |
ALL, acute lymphoblastic leukemia.
We had follow-up data on all 45 patients who participated in the Nordic 2017 rituximab-bendamustine trial.31 On reevaluation, 35 patients (78%) were found to have responded: 24 patients (53%) had achieved CR and 11 (24%) PR. Forty (89%) of all 45 participants were still alive at the final data collection. Figure 4 shows estimated response duration. Median estimated response duration was not reached after 88 months, and estimated 5-year sustained remission was 77%. Table 4 shows frequencies of late-occurring malignancies.
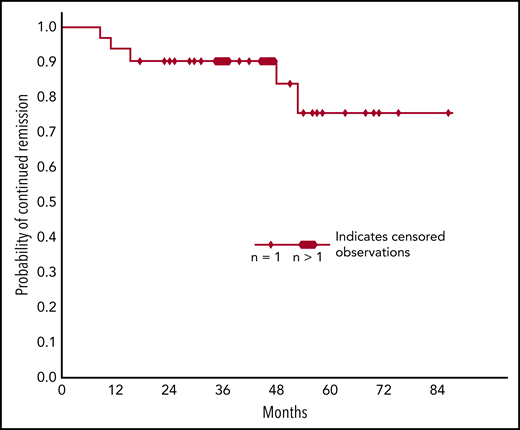
Probability of sustained remission in patients who have responded to 4 cycles of rituximab plus bendamustine. Kaplan-Meier plot. Median estimated response duration is not reached after 88 months and the estimated 5-year sustained response rate is 77%.
Probability of sustained remission in patients who have responded to 4 cycles of rituximab plus bendamustine. Kaplan-Meier plot. Median estimated response duration is not reached after 88 months and the estimated 5-year sustained response rate is 77%.
Response rates following B-cell-directed therapies did not correlate with age, Hb level, or clinical phenotype. In 103 patients with a histologically confirmed clonal LPD and sufficient available data, 87 (84.5%) had responded to 1 or more of B-cell-directed therapies. Among 43 treated patients in whom the bone marrow did not show any definite LPD by histology, the corresponding figures were 32 (74.5%). The difference was not statistically significant.
Discussion
The prevalence and incidence were 4 times higher in Norway compared with Lombardy, Northern Italy, even though the rate of detection of symptomatic cases was probably very high in both populations.6,25,45,46 Unintended patient selection or genetic differences cannot be completely ruled out as an explanation for these discrepancies, which are, however, more likely to be explained by the climatic differences. Most likely, the 7°C lower outdoor temperature in representative parts of Norway compared with Lombardy is associated with more symptomatic disease, resulting in a higher proportion of patients being diagnosed. This first-ever demonstration of a marked association between climate and prevalence and incidence of CAD diagnosis is consistent with the seasonal variations in hemolytic anemia described in individual patients.19-21
We found higher Hb levels than observed in previously published cohorts,6,7 which is probably explained by low selection bias. Nonetheless, anemia was severe (defined as Hb <8 g/dL)1 in 25% to 30% of patients with CAD. Our data do not support a general recommendation of folic acid supplements in CAD,1,3,47 although the serum folate levels might have been influenced by hemolysis.
The clonal bone marrow LPD in CAD was previously perceived as being heterogeneous, classified as a variety of indolent non-Hodgkin lymphomas.6,7 The concept of “CAD-associated LPD” was coined in 2014, based on a systematic histopathology study and later supported by other studies.12,48 Figure 1 shows that many patients diagnosed with other low-grade LPDs (or even no signs of clonal LPD) have later been reclassified as having CAD-associated LPD. In particular, the absence of the MYD88 L265P mutation supports our view that many CAD patients who were previously diagnosed with lymphoplasmacytic lymphoma have been misclassified.12,48,49 Therefore, centralized examination of bone marrow biopsies should be encouraged even in routine clinical practice to optimize diagnostic workup and enable wider recognition of the CAD-associated LPD entity by hematopathologists.
The 16-year estimated survival was longer than previously reported,6,7,18 and 5-year survival was significantly higher than calculated in the registry-based Danish study (83% vs 61%). By comparison, the expected survival in the Norwegian general population is 18.4 years for people (men and women combined) who are already 68 (the median age of CAD diagnosis).50 Despite the lack of matched controls, the strengths of our study are the unselected, large cohort and the stringent diagnostic criteria. We conclude, therefore, that patients with CAD have only a slightly increased mortality. Furthermore, Figure 2 does not confirm a higher death rate during the first 5 years as previously reported.18
Based on age-specific incidences of VTE calculated in a large general population,51 the expected number of VTEs during 8 years in our cohort would be 8.4, whereas we found 14 events. Regarding ATEs, the expected numbers of stroke and MI during 8 years in 232 subjects aged 68 to 76 years should be 13.8 and 11.1, respectively,52,53 whereas we observed 12 strokes and 10 MIs. Drawing conclusions from these observations has some limitations: the retrospective study design, the lack of matched controls, and the low absolute numbers of TEs. Still, our data are consistent with the slightly increased risk of VTEs observed in 2 registry-based studies,17,18 whereas we could not confirm an increased risk of MI or stroke. This study does not support routine use of anticoagulant prophylaxis in patients with CAD. Our results indicate that iron overload is reaching levels that are usually considered to carry a risk of organ damage in one-fifth of subjects with CAD, in particular among those with a history of transfusions.54 The actual clinical consequences remain unclear.
In the 8 patients who developed DLBCL, this retrospective study does not allow conclusions at the individual level regarding clonal relationship with CAD-associated LPD (true transformation) vs de novo DLBCL. However, the expected number of new DLBCL cases in 232 subjects during 8 years can be calculated to 0.13,55 strongly indicating that most DLBCL cases are related to the underlying low-grade LPD. In any case, this 3.4% 8-year risk of transformation is very low.
In this real-life study, we found high response rates and long response duration following therapy with rituximab plus bendamustine or fludarabine. A high proportion of the responses were complete according to strict criteria. Comparison between different studies should be undertaken with caution; however, the inclusion and outcomes criteria were identical and the demographic and baseline characteristics were very similar in the 2 studies addressed. With this reservation, the follow-up data indicate even more favorable response duration with rituximab-bendamustine (median estimated response duration, >88 months; 77% 5-year response duration) than with rituximab-fludarabine. The overall response rates (78% vs 62%) and CR rates (53% vs 38%) were also higher with rituximab-bendamustine. Interestingly, the curves in Figures 3 and 4 seem to reach a plateau after 54 to 80 months, suggesting that a subset of responders will achieve very long-lasting responses and some may even be cured.
It may seem surprising that this study found a higher overall response rate for bendamustine-rituximab than reported in the original prospective trial.31 This can be explained, however, by the long time to response (TTR) and time to best response. In the prospective trial, median TTR was 1.9 months (range, 0.25-10) and time to best response 7.0 months (range, 1.5-30). Hb levels further improved after achievement of response in most responders during the median observation time of 36 months (range, 3-65).31 In this follow-up study, late responses were seen in 3 patients (7% of 45) who previously had been classified as nonresponders in the first study, with a maximum TTR of 24 months; median observation time from treatment was 48 months (range, 9-88). A very late response was even observed in a patient who had been treated with rituximab plus fludarabine. Of note, we also observed a shift toward deeper responses with time following both treatments. A similar occurrence of delayed responses has been reported in Waldenström macroglobulinemia.56 The phenomenon is probably best explained by the existence of long-lived plasma cells that are not able to proliferate and are more or less insensitive to chemoimmunotherapy.57,58 In the 2017 trial, patients received fixed duration 4-cycle therapy.31 The frequently long TTR and improvement of responses with time supports discontinuation after 4 cycles irrespective of remission status.
We found a trend toward more frequent late-occurring malignancies in patients who had participated in the rituximab plus fludarabine trial compared with those who had received rituximab plus bendamustine (Table 4). The difference was not statistically significant, which may be due to the low absolute number of malignancies in each subgroup. Together with the short-time toxicity profiles observed in the prospective trials,31,32 these observations indicate that the rituximab-bendamustine regimen is less toxic than rituximab-fludarabine.
Rituximab monotherapy produced response rates of the same order of magnitude as seen in prospective trials, but the observed median response duration was longer than previously reported.6,29,30 Furthermore, repeat rituximab therapy in relapsed patients will have a fair chance to succeed. Our observation of response to ibrutinib in a single patient does not allow generalization, and a systematic study is warranted.1,3,59
The high efficacy of the B-cell-directed therapies applies to patients with hemolytic anemia as their predominant disease manifestation as well as those with marked ischemic symptoms and mild or compensated hemolysis. According to our data, histologic demonstration of an LPD should not be considered a prerequisite for B-cell-directed therapy, although diagnostic workup should always include a bone marrow biopsy in studies as well as clinical practice.1,3 Age, clinical phenotype, or severity of anemia do not predict response to therapy.
The high percentage of patients who received therapy should be considered with respect to hematologic indications, which showed percentages of mild (or even compensated), moderate, and severe anemia of 38%, 35%, and 27%, respectively. This indicates that some patients with mild anemia are being treated, which may be reasonable because of bothersome circulatory symptoms or fatigue.6,39 This study does not allow any assessment of treatment effect on survival.
In summary, therapy with either rituximab plus bendamustine or rituximab plus fludarabine yields high response rates, high CR rates, and long response durations. These figures appear to be even more favorable for rituximab-bendamustine than for rituximab-fludarabine, and the first combination seems less toxic. The long TTR may be a disadvantage of both regimens. Based on the balance between efficacy, short-term toxicity, and risk of long-term adverse effects, our results show that the use of rituximab plus bendamustine is safe and highly efficacious in relatively fit patients who require therapy for CAD.
Qualified researchers may request nonconfidential original data from the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Astrid Bergrem (Oslo, Norway), Henrik Birgens and Jytte Koch (Copenhagen, Norway), Odd Kildahl-Andersen (Harstad, Norway), Arne Loraas (Skien, Norway), Jakob Nordberg Nørgaard (Tønsberg, Norway), Aud S. Thoresen (Gjøvik, Norway), and Anders Vik (Tromsø, Norway) for providing patient data.
This study was supported by a grant from Western Norway Regional Health Authority (Helse Vest RHF), for which the authors are very grateful.
Authorship
Contribution: S.B. and G.E.T. conceived the idea and wrote the protocol together with W.B., U.R., and S.D.; S.B. analyzed the data and drafted the manuscript in cooperation with W.B., U.R., S.D., and G.E.T; and all authors collected data, discussed the article preparation, reviewed the paper, and approved the submitted version.
Conflict-of-interest disclosure: Outside of this work, S.B. has received research support from Mundipharma; lecture honoraria from Apellis, Bioverativ (a Sanofi company), Janssen-Cilag, and True North Therapeutics; and consultancy and advisory board honoraria from Apellis, Bioverativ, and Momenta Pharmaceuticals. W.B. has received consultancy honoraria and advisory board honoraria from Agios, Alexion, Apellis, Bioverativ (a Sanofi company), Momenta, and Novartis. S.D. has received lecture honoraria from Amgen, Janssen, and Novartis; advisory board honoraria from Sanofi; and grant funding from Janssen and BeiGene. T.H.A.T. has received honoraria from Alexion, Novartis, and Sanofi. B.F. reports consultancies for Apellis, Momenta, and Alexion Pharmaceuticals. A.E.A.D. has received grants from Pfizer AS and personal fees from Bristol-Mayer Squibb, Bayer, Novartis Norway AS, and Pfizer AS. H.F. reports lecture honoraria from Bristol-Meyers-Squibb and Pfizer, and consultancy honoraria from Amgen. G.E.T. has received research support from Mundipharma and lecture honoraria from Mundipharma, AbbVie, Janssen-Cilag, Alexion Pharmaceuticals, and Roche Norway. The remaining authors declare no competing financial interests.
Correspondence: Sigbjørn Berentsen, Department of Research and Innovation, Haugesund Hospital, PO Box 2170, 5504 Haugesund, Norway; e-mail: sigbjorn.berentsen@haugnett.no.
