

In this issue of Blood, illustrate the functional mechanism that determines a megakaryocyte-specific increase in PLAU expression and consequent platelet-dependent fibrinolysis in Quebec platelet disorder (QPD).1
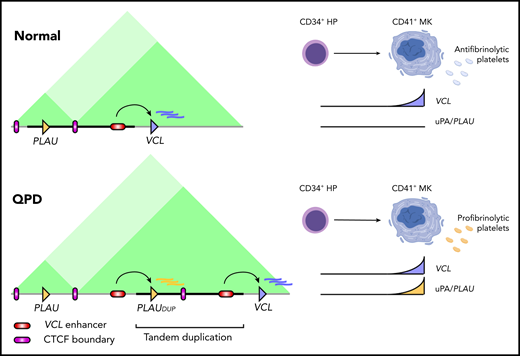
Schematic representation of the interactions across the PLAU locus in nonaffected individuals (normal, top) and in individuals affected by Quebec platelet disorder (QPD, bottom). The different genomic compartments are represented by the pink (sub-TADPLAU) and green box (sub-TADVCL ). The duplicated segments are represented in blue and orange. TAD, topologically associating domain. See Figure 6 in the article by Liang et al that begins on page 2679.
Schematic representation of the interactions across the PLAU locus in nonaffected individuals (normal, top) and in individuals affected by Quebec platelet disorder (QPD, bottom). The different genomic compartments are represented by the pink (sub-TADPLAU) and green box (sub-TADVCL ). The duplicated segments are represented in blue and orange. TAD, topologically associating domain. See Figure 6 in the article by Liang et al that begins on page 2679.
A decade after the discovery that QPD is caused by a tandem duplication,2 Liang et al demonstrate that this causes the additional copy of PLAU to be on the opposite side of a CTCF genomic boundary. As a consequence, the extra copy is not insulated from the effects of one of the transcriptional enhancers of the VCL gene, resulting in a high level of transcription (see figure). This phenomenon is known as “enhancer hijacking” and was observed in Burkitt lymphoma almost 40 years ago.3 In the intervening years, other examples have been discovered, but our ability to diagnose the consequences of new occurrences remains limited and time consuming.
The advent of next-generation sequencing allowed for parallel testing of known causative variants in protein coding genes via gene panels or whole-exome sequencing,3,4 with a diagnostic rate of ∼50% in selected cases of platelet and bleeding disorders. Whole-genome sequencing has begun to show the role of the noncoding portion of the genome in rare diseases,5 but it has also put our as yet incomplete understanding of the regulatory grammar in the human genome under the spotlight. Moreover, the exponentially increasing amount of available whole-genome sequencing has brought to light a large numbers of rare variants of unknown significance, including insertions, deletions, and rearrangements.5
Efforts to catalog the regulatory elements of blood cells6,7 have been largely successful in determining which elements in the genome are marked as active and in which cell type. These findings, coupled with DNA long-range interaction maps,8 have allowed the creation of references for each gene and its regulatory landscape. These have been extremely useful for pinpointing causal genetic variants,5 but it is still difficult to draw general conclusions that can be applied each time a genetic variant is suspected to be responsible for a disease. Our understanding of buffering and compensatory mechanisms between regulatory sequences of gene expression thresholds and of genome compartmentalization is still at a stage at which multiple lines of evidence are required to unequivocally assign causality to noncoding genetic variants and to determine the mechanism of action. Liang et al used a robust study design involving a combination of recall by genotype, publicly available data, an animal model, and ex vivo molecular biology experiments. Altogether, the evidence demonstrated that the tandem duplication previously associated with QPD places the additional copy of PLAU in front of a strong transcriptional enhancer, which under normal conditions, drives the expression of the VCL gene to high levels but is restricted from accessing PLAU because of the DNA local 3-dimensional conformation and the boundary between the two. The VCL enhancer is also part of the duplication; therefore, VCL expression remains unaffected and the additional CTCF site present does not create additional DNA loops. In summary, this study nicely illustrates the necessary steps to ascertain the causal role of noncoding genetic variants and their mechanism of action in the setting of hematologic rare diseases when there are only a handful of affected individuals worldwide.
Conflict-of-interest disclosure: The author declares no competing financial interests.
