


Abstract
The treatment of multiple myeloma (MM) is currently being redefined by humoral and cellular immunotherapies. For decades, there was limited belief in immune-based anti-MM therapy as a result of the moderate graft-versus-myeloma effect of allogeneic stem cell transplantation. Today, monoclonal antibodies comprise the new backbone of anti-MM therapy, and T-cell therapies targeting BCMA are emerging as the most potent single agents for MM treatment. Herein, we present our assessment of and vision for MM immunotherapy in the short and midterm.
The future is now: Ab immunotherapy is the new backbone of MM therapy
Since the approval of combination therapies containing monoclonal antibodies (mAbs) for patients with relapsed multiple myeloma (MM), the anti-CD38 mAb daratumumab (Dara) has become the new backbone of first-line therapy in transplantation-eligible and -ineligible MM patients. In the CASSIOPEIA trial, a quadruple-drug regimen of Dara plus bortezomib, thalidomide, and dexamethasone (Rd; Dara-VTD) led to an overall response rate (ORR) of 93% and progression-free survival (PFS) rate of 93%, vs 90% and 85% in the control arm at 18 months, respectively, setting a new benchmark for efficacy in induction therapy.1 Minimal residual disease (MRD) negativity was achieved in 64% of patients, vs 44% in the VTD control arm, suggesting improved overall survival (OS) with longer follow-up. On the basis of these results, the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved Dara-VTD in early 2020.
Likewise, the induction regimen Dara plus bortezomib, lenalidomide, and dexamethasone (Dara-VRD), containing lenalidomide instead of thalidomide, achieved compelling results in the GRIFFIN phase 2 study, with PFS and OS rates of ≥95% at 24 months.2 Notably, PFS rates were not significantly different between Dara-VRD and VRD, but there was a marked difference in MRD negativity in the Dara arm (51.0% vs 20.4% at 22 months), which will likely translate into better PFS with longer follow-up.2 The phase 3 study PERSEUS of Dara-VRD vs VRD has completed enrollment, and results are expected to be reported in 2022. In transplantation-ineligible MM patients, combination therapies that include Dara as backbone (eg, Dara plus bortezomib, melphalan, and prednisone [Dara-VMP]3 and Dara plus Rd [Dara-Rd]4 ) have recently been approved. Subcutaneous injection of Dara5 was recently approved, and accordingly, Dara-VMP and Dara-Rd can be considered the new standards for transplantation-ineligible patients with newly diagnosed (ND) MM.
Building on the successful clinical use of Dara, alternative anti-CD38 Abs are taking the stage, with isatuximab (Isa) being the most clinically advanced candidate. In the randomized IKARIA trial, the triple-drug regimen of Isa, pomalidomide, and Rd was superior to control treatment with pomalidomide plus Rd in relapsed/refractory (RR) MM patients, with PFS of 11.5 and 6.5 months, respectively.6 On the basis of this study, the FDA approved this regimen in March 2020, and we anticipate EMA approval in 2020 as well. Like the PERSEUS trial, the GMMG HD7 trial is evaluating induction therapy with Isa-VRD vs VRD in a randomized design. It is likely that both anti-CD38 Abs (ie, Dara and Isa) will be approved in combination regimens for use in first-line therapy. Numerous additional regimens with anti-CD38 Abs (eg, in combination with carfilzomib7,8 or traditional chemotherapy9 ) are being evaluated and will enrich the therapeutic armamentarium to achieve disease control in RR MM patients.
The primary modes of action of Dara and Isa are distinct; however, the activity of both mAbs is in part dependent on the density of CD38 molecules along the myeloma cell membrane.10 Accordingly, strategies to increase CD38 expression on MM cells (eg, by epigenetic modulation) are being investigated.11,12 Of note, Dara also depletes CD38+ immunosuppressive cells, which is associated with an increase in cytotoxic T cells, potentially contributing to the activity of this Ab.13 An unresolved question is whether prior therapy with an anti-CD38 mAb affects the subsequent efficacy of T cell–redirecting therapies. Because CD38 is expressed on activated T cells (and to a lesser extent on resting T cells and natural killer [NK] cells), anti-CD38 mAbs may lead to pertubations in T-cell composition and may interfere with the modes of action of chimeric antigen receptor (CAR) T-cell therapies and T cell–engaging bispecific Abs (bsAbs). Careful investigations are warranted to determine the optimal sequences and timings of anti-CD38 and T cell–redirecting therapies.
The role of elotuzumab (Elo), an anti-SLAMF7 mAb, in MM therapy is less clear. The anti-MM potency of Elo as a single agent is rather limited, and therefore, Elo has been evaluated in combination with immune-modulating agents to augment activity. Recently, final data from the ELOQUENT-1 trial, which evaluated Elo, lenalidomide, and Rd (Elo-Rd) vs Rd in transplantation-ineligible ND MM patients, did not demonstrate additive activity of Elo.14 However, in the relapsed setting, Elo-Rd was superior to the Rd control arm.15 Furthermore, Elo in combination with pomalidomide and Rd exerted significant clinical activity in the ELOQUENT-3 study, with 40% of RR MM patients being in remission at 2 years.16 Given that Elo is well tolerated and has a favorable safety profile, this Ab may also play a role in the setting of maintenance therapy. Currently, Elo is being evaluated in quadruple-drug induction and consolidation regimens in transplantation-eligible patients with ND MM (HD6 trial, Elo-VRD; DSMMXVII trial, Elo plus carfilzomib, lenalidomide, and Rd). Notably, T cells expressing an SLAMF7 CAR with a targeting domain derived from Elo are substantially more potent against MM than Elo in preclinical models in vitro and in vivo,17 and therefore, the results of phase 1/2A clinical trials with SLAMF7 CAR T cells (CARAMBA and MELANI-01 trials) are eagerly awaited.
A new future begins: the dawn of T-cell–redirecting therapies
CAR T cells
T cell–redirecting therapies with genetically engineered CAR T cells and T-cell–engaging bsAbs currently comprise the most exciting new developments in cancer immunotherapy and will change the treatment paradigm for MM (Figure 1). Idecabtagene-vicleucel (Ide-cel; bb2121) is a BCMA CAR T-cell therapy for which an ORR of 85% was reported, with 45% of participants achieving complete response (CR) in a dose-escalating phase 1 study of heavily pretreated RR MM patients.18 Full recruitment has been achieved in the pivotal phase 2 KarMMa study, and initial results were presented at the 2020 ASCO meeting, showing an ORR of 73% and median PFS of 8.6 months, both of which increased with higher doses.19 Therefore, we are expecting FDA approval of Ide-cel for RR MM in 2020, which will constitute a landmark as the first genetically engineered T-cell therapy in MM.
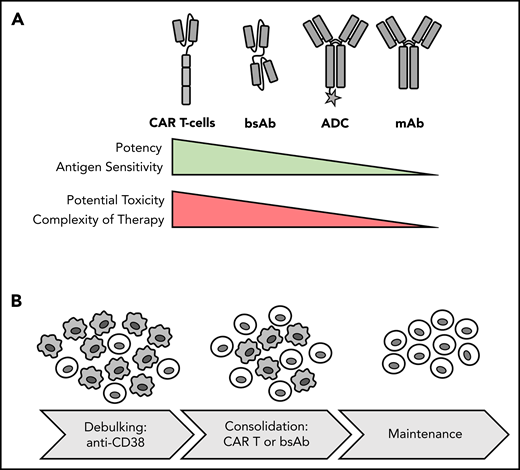
Potency, toxicity and sequencing of different forms of immunotherapy. (A) Comparison of immunotherapy treatment modalities. (B) Potential therapeutic sequence for newly diagnosed MM: debulking with anti-CD38 Ab-based regimen and consolidation and induction of MRD− status with T-cell–redirecting therapy, such as CAR T-cell therapy, followed by maintenance. Gray cells indicate MM cells; light cells indicate T cells.
Potency, toxicity and sequencing of different forms of immunotherapy. (A) Comparison of immunotherapy treatment modalities. (B) Potential therapeutic sequence for newly diagnosed MM: debulking with anti-CD38 Ab-based regimen and consolidation and induction of MRD− status with T-cell–redirecting therapy, such as CAR T-cell therapy, followed by maintenance. Gray cells indicate MM cells; light cells indicate T cells.
LCAR-B38M (also known as JNJ-68284528) is another compelling BCMA CAR T-cell product currently under investigation in phase 1/2 trials.20 Interim results from the CARTITUDE-1 study showed an ORR of 100%, with 76% of heavily pretreated RR MM patients achieving CR.21 Several other BCMA CAR T-cell products are being investigated in phase 1 studies, and collectively, the data support the notion that BCMA CAR T cells are highly effective and probably constitute the most potent single agent available in the RR disease setting.18,22,23 However, duration of response has been limited in these trials, and a majority of patients have ultimately relapsed. In contrast to CD19 CAR T cells in non-Hodgkin lymphoma,24 there is no survival plateau in MM so far.
Overall, BCMA CAR T-cell therapy has displayed a favorable safety profile, with lower incidence of cytokine release syndrome and neurotoxicity, compared with CD19 CAR T-cell therapy in B-cell leukemia and lymphoma.18,25 However, the clinical experience with BCMA CAR T cells has also exposed several challenges associated with targeting this antigen, and potential mechanisms of relapse or resistance include antigen downregulation or even loss26 in a small subset of patients, as well as limited persistence of BCMA CAR T cells and limited fitness of T cells in heavily pretreated patients.18,20,22,23,27 Several strategies are being pursued to address these challenges, including the use of γ-secretase inhibitors to enhance BCMA molecule density on MM cells and reduce the amount of soluble BCMA in serum,28 refined CAR T-cell manufacturing protocols to increase fitness (eg, in the presence of PI3K inhibitors),29 and use of CAR products with defined T-cell subset compositions and humanized targeting domains to reduce immunogenicity and promote engraftment and in vivo expansion.28,30,31
ADCs
A subset of MM patients with significant comorbidities may be less able to tolerate the potential toxicity associated with T-cell–redirecting therapies, and for this patient population, BCMA-specific Ab-drug conjugates (ADCs) may be an alternative. A lead candidate in this class of compounds is belantamab mafadotin. Belantamab mafadotin eliminates MM cells by releasing the cytotoxic agent auristatin F and through Ab-dependent cellular cytotoxicity. Belantamab mafadotin is administered IV every 3 weeks and is well tolerated, except for corneal toxicity, which occurs in >70% of patients.32 The ORR with belantamab mafadotin was ∼30% in a phase 2 trial of patients refractory to Dara, immunomodulatory imide drugs (IMiDs), and proteasome inhibitors.33 PFS durations were 2.8 and 3.9 months in the 2.5 and 3.4 mg/kg dose groups, respectively. In responding patients, PFS times were not reached and 8.4 months in the 2.5 and 3.4 mg/kg dose groups, respectively.34 On the basis of these results, we anticipate FDA approval in triple-refractory RR MM later in 2020. Intriguingly, the mode of action of belantamab mafadotin is independent of T-cell fitness, and accordingly, this ADC is given consideration as one of the few remaining therapeutic options for patients who experience relapse after CAR T-cell therapy.35
The future ahead: a race between BCMA-targeting agents to first-line therapy?
T-cell–engaging bsAbs are another way to harness the power of a cellular immune response to combat MM (Figure 1). A first proof of concept for bsAbs in MM was recently provided with AMG420, a CD3×BCMA bispecific T-cell engager construct. At a dose of 400 μg per day, the response rate was 70%, including 50% MRD− CRs, with some responses lasting >1 year.36 Recently, a pilot study of the asymmetric CD3×BCMA bsAb CC-9326937 reported an 89% ORR and 44% CR rate at the highest dose of 10 mg in heavily pretreated MM patients.38 Teclistamab and REGN5458 are 2 other CD3×BCMA bsAbs showing promising preliminary results in early clinical trials.39,40
A practical advantage of bsAbs over CAR T cells is that they are off-the-shelf products and can be administered repeatedly to sustain the therapeutic pressure against MM. A current focus in the field is to determine the most active bsAb compound that we anticipate will advance to pivotal trials within the next 1 to 3 years and obtain detailed insights into potential mechanisms of resistance to bsAb therapy resulting from interference of soluble BCMA protein, BCMA downregulation, and humoral and cellular immune responses against synthetic bsAb constructs.41
We anticipate that T-cell–redirecting therapies targeting BCMA (eg, CAR T cells, bsAbs) will rapidly move forward from treatment of late-stage RR MM to earlier treatment lines and even to first-line therapy in difficult-to-treat MM patients. A key assumption supporting this strategy is that the fitness of endogenous T cells at early disease stages is higher compared with later disease stages, when patents have received multiple rounds of cytotoxic therapy.42 Although data supporting this hypothesis are still emerging in MM,43 we recently showed in lymphoma that patient T cells acquire functional defects after chemotherapy, which affects activity of bsAbs.44
A challenge to establishing BCMA CAR T cells and bsAbs as first-line therapies is the requirement to demonstrate superiority over standard of care, where Dara-based combinations have now set a high bar in efficacy and safety. Accordingly, several studies are focusing on subgroups of MM patients with suboptimal responses to and outcomes after conventional and Dara-based combination regimens, because these patients may particularly benefit from CAR T-cell and bsAb therapies. The randomized KarMMa 3 study is evaluating Ide-cel vs standard of care in RR MM patients previously treated with Dara, an IMiD, and a proteasome inhibitor who have received ≥2, but not >4 prior regimens. The CARTITUDE-4 study is investigating LCAR-B38M in patients who have received 1 to 3 prior lines of therapy, been preexposed to proteasome inhibitors, and are resistant to lenalidomide. Of note, there are limited safety data on BCMA CAR T-cell therapy in the transplantation-ineligible MM patient population. Similarly, there is a strategy to implement the ADC belantamab mafadotin earlier in MM treatment (eg, the DREAMM 6 study is evaluating belantamab mafadotin in combination with Rd or VRD in second-line treatment). The DREAMM 9 study will evaluate belantamab mafadotin together with standard of care as induction therapy for transplantation-eligible ND MM patients.
Key requirement: biomarkers to guide patient selection and choice of immunotherapy
Identifying suboptimal response to and early relapse after induction therapy and high-dose chemotherapy with subsequent autologous stem cell transplantation45 is another approach to define MM patients who may particularly benefit from CAR T-cell therapy as an element of first-line treatment. An alternative strategy is to identify high-risk MM patients at primary diagnosis using molecular markers, which is not without challenges because of the inter- and intrapatient tumor heterogeneities in MM.46,47 We consider several markers as being potentially suitable for identifying high-risk patients for inclusion in CAR T-cell and bsAb therapy studies, including revised International Staging System stage 3 status,48 a TP53 double-hit event,49,50 gene-expression profiling–defined high-risk status,51,52 and presence of extramedullary MM disease.53,54 The most recent fluorescence in situ hybridization–based MM risk classifier is another promising tool for patient stratification.55 We would consider an OS of ≥3 years in this MM patient subgroup to be a breakthrough that now seems accomplishable in the immunotherapy era.
MRD is another key marker defining a subgroup of MM patients with suboptimal responses and outcomes. The number of MRD+ patients is significant, and such patients constitute ∼40% of ND MM patients after high-dose chemotherapy and autologous stem cell transplantation.1 For these patients, the goal of administering CAR T-cell therapy is conversion to MRD− status with ensuing long-term disease-free survival and even cure. It is important to note that eradication of MM cells in the MRD setting is a prerequisite for cure. Recent data have suggested that a primary mode of action of CAR T cells against hematologic cancer cells is the induction of apoptosis,56 and it remains to be determined whether metabolically inactive MM cells in MRD+ patients can be sufficiently eliminated. Double-hit TP53 lesions are found in 4% of ND MM patients,49 increase in frequency over the course of disease,57,58 and provide another mechanism of apoptosis resistance. A recent study revealed that impaired FAS receptor signaling is associated with failure of CD19 CAR T-cell therapy in acute leukemia.59 It is noteworthy that genes involved in apoptosis induction are frequently altered as a result of mutations and chromosomal aberrations in MM.60,61
Burning questions and outlook: the future is bright for immunotherapy in MM
There are several burning questions centered around identifying biomarkers that predict outcome and identifying patients who have the highest chance of benefiting from T-cell–redirecting therapies, identifying the optimal single antigen or combination of antigens for T-cell–redirecting therapies to consistently induce durable complete remissions, and determining whether eventually MM can be cured in a relevant subset (or even a majority) of patients (Table 1). At our institution, we have treated MM patients with biallelic TP53 inactivation and very aggressive disease and observed rapid and deep responses after BCMA CAR T-cell therapy. This is in line with data from, for example, the KarMMa study, where a majority of patients with high-risk cytogenetics and extramedullary disease responded to CAR T-cell treatment.18 These data suggest that current algorithms for staging and risk assessment in MM should be adapted in the immunotherapy era.
Top 10 burning questions for immunotherapy in MM
| Question | Assessment and perspective in 2020 |
|---|---|
| Will high-risk disease become standard risk in the era of novel CAR T-cell therapy? | Potentially yes. BCMA CAR T cells are effective in patients with high-risk cytogenetics, including double-hit TP53 mutation. |
| Is MRD negativity the best end point for immunotherapy trials? | Probably yes. However, there is an issue with obtaining high-quality MRD samples shortly after CAR T-cell therapy because of hypocellular bone marrow. Furthermore, MRD assessment should be combined with functional imaging to exclude residual focal lesions or extramedullary disease. |
| Should we deliver novel immunotherapies preferentially in early treatment lines (and what is the optimal sequence of novel immunotherapies)? | Probably yes. T cells from patients in early disease stages and early in the treatment course exhibit better fitness compared with those in late stages. Determining the optimal sequence of immunotherapy modalities is an open issue (eg, What is the optimal interval between Dara and Elo [because Dara diminishes NK cells that are required for Elo mode of action? If 1 BCMA-targeting agent fails, can another still work? How soon after CAR T-cell therapy can bsAbs be considered [if lymphopenia after lymphodepletion has not resolved]?). |
| Is relapse from immunotherapy more difficult to treat? | Probably no. However, the selective pressure of immunotherapies on tumor and microenvironment has yet to be defined. |
| Will we cure myeloma with CAR T cells and T cell–engaging bsAbs? | Probably yes. A subgroup of patients with low tumor burden and favorable biology will exceptionally benefit. |
| Will we deliver chemotherapy-free immunotherapy combinations? | Probably yes. Strategies include combination of anti-CD38 mAbs with ADCs or bsAbs (anti-BCMA or anti-GPRC5D) or combination of T-cell–redirecting therapies with IMiDs to reprogram and improve fitness of endogenous T cells. |
| Do we need additional diagnostics in the era of immunotherapy? | Probably yes. There is a need and opportunity to implement advanced diagnostics in MM immunotherapy to guide patient and antigen selection and monitor disease response as well as advanced genomic analyses to monitor clonal composition and evolution and potential gene or even chromosome loss. |
| Will targeting >1 antigen on MM cells improve response rates and durability of response? | Probably yes. Multiple-antigen targeting is an appealing strategy to counteract the antigen downregulation and loss that limit the efficacy of BCMA CAR T cells in a subset of patients in current clinical trials. |
| Is there a place for immunotherapy in early asymptomatic stages of MM? | Probably yes, but these therapies need to be well tolerated. |
| Will the COVID-19 pandemic stop the success of novel immunotherapies? | Definitely no, but the timelines for completion of trials, approval of novel trials, and reimbursement may decelerate. |
| Question | Assessment and perspective in 2020 |
|---|---|
| Will high-risk disease become standard risk in the era of novel CAR T-cell therapy? | Potentially yes. BCMA CAR T cells are effective in patients with high-risk cytogenetics, including double-hit TP53 mutation. |
| Is MRD negativity the best end point for immunotherapy trials? | Probably yes. However, there is an issue with obtaining high-quality MRD samples shortly after CAR T-cell therapy because of hypocellular bone marrow. Furthermore, MRD assessment should be combined with functional imaging to exclude residual focal lesions or extramedullary disease. |
| Should we deliver novel immunotherapies preferentially in early treatment lines (and what is the optimal sequence of novel immunotherapies)? | Probably yes. T cells from patients in early disease stages and early in the treatment course exhibit better fitness compared with those in late stages. Determining the optimal sequence of immunotherapy modalities is an open issue (eg, What is the optimal interval between Dara and Elo [because Dara diminishes NK cells that are required for Elo mode of action? If 1 BCMA-targeting agent fails, can another still work? How soon after CAR T-cell therapy can bsAbs be considered [if lymphopenia after lymphodepletion has not resolved]?). |
| Is relapse from immunotherapy more difficult to treat? | Probably no. However, the selective pressure of immunotherapies on tumor and microenvironment has yet to be defined. |
| Will we cure myeloma with CAR T cells and T cell–engaging bsAbs? | Probably yes. A subgroup of patients with low tumor burden and favorable biology will exceptionally benefit. |
| Will we deliver chemotherapy-free immunotherapy combinations? | Probably yes. Strategies include combination of anti-CD38 mAbs with ADCs or bsAbs (anti-BCMA or anti-GPRC5D) or combination of T-cell–redirecting therapies with IMiDs to reprogram and improve fitness of endogenous T cells. |
| Do we need additional diagnostics in the era of immunotherapy? | Probably yes. There is a need and opportunity to implement advanced diagnostics in MM immunotherapy to guide patient and antigen selection and monitor disease response as well as advanced genomic analyses to monitor clonal composition and evolution and potential gene or even chromosome loss. |
| Will targeting >1 antigen on MM cells improve response rates and durability of response? | Probably yes. Multiple-antigen targeting is an appealing strategy to counteract the antigen downregulation and loss that limit the efficacy of BCMA CAR T cells in a subset of patients in current clinical trials. |
| Is there a place for immunotherapy in early asymptomatic stages of MM? | Probably yes, but these therapies need to be well tolerated. |
| Will the COVID-19 pandemic stop the success of novel immunotherapies? | Definitely no, but the timelines for completion of trials, approval of novel trials, and reimbursement may decelerate. |
There is a rich pipeline of novel CAR T-cell products, bsAbs, and trispecific Abs62 that target alternative antigens, including SLAMF7,17 CD44v6,63 and GPRC5D,64 as well as multispecific CAR T cells.65 Indeed, it will be important to determine whether targeting 2 (or more) antigens can exert more therapeutic pressure to counteract downregulation of a single antigen and clonal evolution of MM cells. In another approach, NK cells are used as effector cells; CAR-modified NK cells as well as bispecific killer cell engager–targeting MM antigens have shown encouraging results in preclinical studies.66,67 A recent report on a phase 1/2 trial evaluating CD19 CAR NK cells in patients with non-Hodgkin lymphoma and chronic lymphocytic leukemia showed a high rate of initial responses and a favorable toxicity profile.68 We recently demonstrated that advanced microscopic techniques can be used to identify and monitor target antigens on MM cells to inform therapeutic decisions.69 Significant efforts are being undertaken to simplify the manufacturing and logistics around CAR T-cell therapy involving virus-free gene transfer, automated point-of-care production, and allogeneic cell products.70,71 With these developments, we are confident that the role of immunotherapy in MM will be manifested, and the prospect of a chemotherapy-free free yet curative treatment for a majority of MM patients can become real within the next decade.
Acknowledgments
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, project #324392634, TRR 221; M.H. and H.E.), the German Ministry for Education and Research (Bundesministerium für Bildung und Forschung, project IMMUNOQUANT; M.H. and H.E.), the EU Horizon 2020 research and innovation program under grant agreements 733297 (EURE-CART; M.H. and H.E.) and 754658 (CARAMBA; M.H. and H.E.), the Myeloma Crowd Research Initiative (M.H. and H.E.), and patient advocacy groups Hilfe im Kampf gegen den Krebs e.V. (Würzburg, Germany) and Forschung hilft–Stiftung zur Förderung der Krebsforschung an der Universität Würzburg (L.R., M.H., and H.E.). L.R. is supported by German Cancer Aid (Deutsche Krebshilfe e.V.) as a fellow in the Mildred Scheel Early Career Center Würzburg (Mildred Scheel Nachwuchzentrum).
Authorship
Contribution: L.R., M.H., and H.E. wrote and approved the manuscript.
Conflict-of-interest disclosure: L.R. has participated in scientific advisory boards for Janssen, Celgene/Bristol-Myers Squibb, GlaxoSmithKline, and Sanofi and has received research support from Skyline Dx. M.H. has participated in scientific advisory boards for Janssen and Celgene/Bristol-Myers Squibb and is listed as an inventor on patent applications related to CAR technologies that have been filed by the University of Würzburg. H.E. has participated in scientific advisory boards for Janssen, Celgene/Bristol-Myers Squibb, Amgen, Novartis, and Takeda; has received research support from Janssen, Celgene/Bristol-Myers Squibb, Amgen, and Novartis; and has received honoraria from Janssen, Celgene/Bristol-Myers Squibb, Amgen, Novartis, and Takeda.
Correspondence: Hermann Einsele, Universitätsklinikum Würzburg, Medizinische Klinik und Poliklinik II, Oberdürrbacherstrasse 6, 97080 Würzburg, Germany; e-mail: einsele_h@ukw.de.
