

This issue of Blood presents a tour de force: 1 report a phase 1/2 trial of zanubrutinib, a novel Bruton tyrosine kinase (BTK) inhibitor, and lay the groundwork for .2 to present a head-to-head comparison of 2 BTK inhibitors, zanubrutinib and ibrutinib, in MYD88L256P mutant Waldenström macroglobulinemia (WM). These companion articles on zanubrutinib will arguably have broad implications in WM therapeutics. The 2 trials showcase how extraordinary international collaborative efforts were instrumental in the development of zanubrutinib at warp speed for treatment of a rare B-cell malignancy.
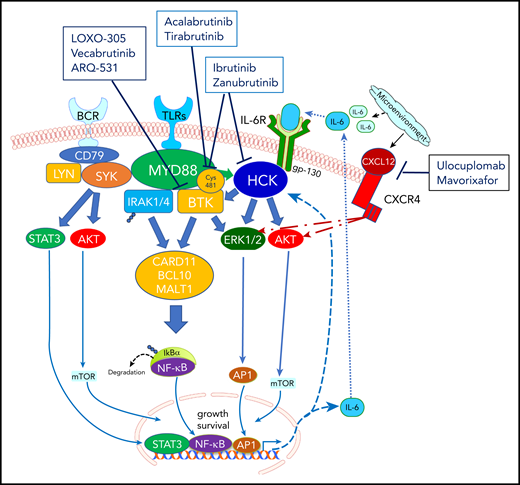
Prosurvival signaling triggered by mutated MYD88 in WM and its interaction with BTK. BTK, a protein that is uniquely positioned in the B-cell receptor (BCR) signal transduction pathway, upon phosphorylation, interacts with myeloid differentiation primary response protein (MYD88), a Toll-like receptor (TLR) signaling protein, leading to an adaptive immune response, with IgM formation. BTK inhibition reduces MYD88-BTK complexing, with resultant downregulation of NFκB and apoptosis of WM cells. Both ibrutinib and zanubrutinib irreversibly attach via covalent bonding to Cys481 residue within the BTK pocket, permanently shutting off its signaling capability by disrupting ATP binding. In addition, both ibrutinib and zanubrutinib target HCK, a SRC family member responsible for activating BTK in response to mutated MYD88. Two other irreversible BTK inhibitors, acalabrutinib and tirabrutinib, have demonstrated efficacy in WM. In addition, reversible BTK inhibitors, including vecabrutinib, LOXO-305, and ARQ 531, which can bind noncovalently to BTK, are under evaluation, given their potential to dually inhibit wild-type BTK and Cys481S-mutated BTK. Ulocuplumab, a monoclonal antibody against CXC chemokine receptor 4 (CXCR4) and mavorixafor, a CXCR4 allosteric inhibitor, are being evaluated in partnership with ibrutinib, to overcome CXCR4-mediated resistance.
Prosurvival signaling triggered by mutated MYD88 in WM and its interaction with BTK. BTK, a protein that is uniquely positioned in the B-cell receptor (BCR) signal transduction pathway, upon phosphorylation, interacts with myeloid differentiation primary response protein (MYD88), a Toll-like receptor (TLR) signaling protein, leading to an adaptive immune response, with IgM formation. BTK inhibition reduces MYD88-BTK complexing, with resultant downregulation of NFκB and apoptosis of WM cells. Both ibrutinib and zanubrutinib irreversibly attach via covalent bonding to Cys481 residue within the BTK pocket, permanently shutting off its signaling capability by disrupting ATP binding. In addition, both ibrutinib and zanubrutinib target HCK, a SRC family member responsible for activating BTK in response to mutated MYD88. Two other irreversible BTK inhibitors, acalabrutinib and tirabrutinib, have demonstrated efficacy in WM. In addition, reversible BTK inhibitors, including vecabrutinib, LOXO-305, and ARQ 531, which can bind noncovalently to BTK, are under evaluation, given their potential to dually inhibit wild-type BTK and Cys481S-mutated BTK. Ulocuplumab, a monoclonal antibody against CXC chemokine receptor 4 (CXCR4) and mavorixafor, a CXCR4 allosteric inhibitor, are being evaluated in partnership with ibrutinib, to overcome CXCR4-mediated resistance.
The approval in 2015 of ibrutinib, the first in-class BTK inhibitor for WM, was a watershed moment that followed, in short order, the discovery of a highly prevalent mutation in MYD88 in patients with WM.3 The understanding that mutated MYD88 triggers prosurvival signaling through BTK provided the rationale for investigating BTK inhibitors.4-6 Like ibrutinib, zanubrutinib irreversibly attaches to BTK,7 permanently shutting off its signaling capability (see figure). However, zanubrutinib may offer reduced off-target kinase inhibition, potentially improving tolerability.
Trotman et al evaluated zanubrutinib administered at 2 different doses (160 mg twice daily and 320 mg once daily) until progression, through a phase 1/2 trial (BGB3111-Au003) involving treatment-naive patients (n = 24) and patients with relapsed/refractory WM (n = 53). The overall response rate was 96%, with a respectable subset achieving at least a very good partial response (VGPR). Notably, VGPR rates improved over time, from ∼21% at 6 months to ∼44% at 2 years. Ten patients discontinued zanubrutinib because of treatment-emergent toxicities. The twice-daily dosing schedule that resulted in more sustained BTK inhibition was selected to move forward.
These promising data provided the impetus for the international, phase 3 ASPEN trial through which Tam and colleagues sought to leverage the deeper responses induced by zanubrutinib. Demonstrating the superiority of the magnitude of response over ibrutinib, a standard of care for WM, appeared to be a low-hanging fruit. With a primary endpoint that was somewhat unconventional for a phase 3 study and a robust international partnership, the ASPEN study had the trappings of a trial designed to facilitate brisk drug development in a rare malignancy for which differences in the progression-free survival (PFS), and more so, overall survival (OS), can take a long time to emerge. The primary endpoint was the proportion of patients achieving at least a VGPR as the best response, as assessed by an independent review committee at 12 months after the last patient’s enrollment. With 1:1 randomization, ASPEN expeditiously accrued 199 evaluable WM (treatment naive, n = 37; relapsed/refractory, n = 162) patients. Although complete response (CR) remained elusive (despite administration of drugs in both arms until progression), 28% and 19% achieved VGPR with zanubrutinib and ibrutinib, respectively (P = .09). Major response rates were strikingly similar (77% vs 78%), as were the 18-month PFS rates (84% and 85%) during the short follow-up. Barring grade 3 neutropenia, the toxicity-profile comparisons favored zanubrutinib over ibrutinib, also reflected by a lower discontinuation rate for zanubrutinib.
The ASPEN investigators had set a lofty goal of demonstrating at least a 20% improvement in VGPR/CR rates (35% with zanubrutinib vs 15% with ibrutinib). The justification for keeping VGPR/CR as a primary endpoint was ostensibly the feasibility of early analysis, with rapid attainment of responses and curtailment of the sample size in an uncommon indolent malignancy for which the subjects would have otherwise needed protracted follow-up. Although the high VGPR rates observed with zanubrutinib in the BGB3111-Au-003 trial were noteworthy, it is not known whether the VGPR/CR rates are valid surrogate efficacy endpoints in WM. The magnitude of response, a time-dependent variable, is known to improve with continuous as well as fixed-duration therapy in WM. In the pivotal trial involving ibrutinib, the VGPR rates improved substantially from 15% to 29%, with longer follow-up.6,8 In ASPEN a numerically higher proportion of patients achieved VGPR with zanubrutinib, but the issue at heart is the real-world consequence of this finding. There are currently no prospective data on the use of BTK inhibitors to suggest that attainment of VGPR or deeper responses translates into substantive improvement in clinically meaningful endpoints of PFS and OS. Moreover, the 12-month PFS landmark analysis by best response in BGB3111-Au-003 demonstrably failed to correlate deep response with prolonged PFS. In ASPEN, the control arm performed as expected, mirroring the results of the pivotal trial and reinforcing ibrutinib’s tenacity.6,9 Zanubrutinib performed somewhat below the ambitious expectations built upon the BGB3111-Au-003 data, underscoring the value of conducting phase 3 trials and longer follow-up. In addition, it brought into question excessive reliance on response rate and depth (known to be influenced by the CXCR4 mutation status of patients receiving BTK-targeted therapies)10 as the primary endpoint. Still, a sustained IgM reduction, as was demonstrated in favor of zanubrutinib, may be clinically pertinent, as WM-related morbidity, including peripheral neuropathy, cryoglobulinemia, cold agglutinemia, coexisting A(H)L amyloidosis, and hyperviscosity is driven by the monoclonal IgM protein.11
The key findings of these 2 studies undeniably pull back the curtain on the uniqueness of WM, an IgM lymphoplasmacytic lymphoma,11 with a near-ubiquitous presence of clonal mutated MYD88 molecular signature and subclonal CXCR4 mutations, encountered in 30% to 40% cases.12,13 Both genetic alterations are intricately linked, but have opposing effects on the efficacy of BTK-targeting agents: mutated MYD88 confers sensitivity, whereas CXCR4 mutations confer resistance. An important limitation for ASPEN was the low (9%) detection rate for CXCR4 mutations by Sanger sequencing, a stratification variable in the cohort. This issue was mitigated, in part, with a post hoc analysis, using the more sensitive next-generation sequencing, which increased the detection rate to 28%. More importantly, it unearthed an inequality in the distribution of patients with CXCR4 mutations across the 2 arms, favoring the ibrutinib arm (34% with zanubrutinib vs 22% with ibrutinib), a potential explanation for zanubrutinib narrowly missing the primary endpoint target. Fewer VGPRs were observed in both ibrutinib- and zanubrutinib-treated patients with mutated CXCR4 in comparison with those with wild-type CXCR4.
Although, both drugs demonstrated toxicities attributable to continuous inhibition of kinases, the stark differences in the intensities of adverse effects were exposed with the randomized controlled design of ASPEN.2 Because both were oral agents, an easily adoptable double-blind design, instead of the open-label study, could have reduced biases. Nonetheless, the well-recognized and oft-dreaded cumulative cardiovascular complications of atrial fibrillation (2% vs 15%) and grade 3 hypertension (6% vs 11%) were conspicuously lower with zanubrutinib. Similarly, the rates of diarrhea, muscle spasms, edema, and contusion favored zanubrutinib. However, grade 3 neutropenia (20% vs 8%), occurred more frequently with zanubrutinib, necessitating greater use of granulocyte colony-stimulating factor (47% vs 31%). Whether the profound neutropenia observed with zanubrutinib will pose challenges to partnering it with other agents remains to be seen.
Although the ASPEN study did not meet its primary endpoint, both ASPEN and BGB3111-Au-003 are highly instructive trials, particularly when their findings are put into clinical context. The investigators should be commended for their valuable contribution. In hindsight, should ASPEN trialists have adopted a noninferiority design (with PFS as the primary endpoint), particularly when it is not necessarily the greater degree of BTK inhibition, but the reduced off-target effects (as exemplified by more favorable toxicity-profile and quality-of-life assessments) that is more likely to strike a chord with patients and practicing hematologists alike? Or will once-a-day convenience and longer clinical experience with ibrutinib drive clinical decision making? Regardless, for the pragmatist WM community, zanu zings, and will likely become an important treatment option for patients with WM.
Conflict-of-interest disclosure: P.K. has received research funding and has been a consultant to AbbVie, GlaxoSmithKline, Sanofi, and Takeda; has been a consultant to Cellectar, Karyopharm, and Pharmacyclics; and has received research funding from Amgen. S.P.T. has received research funding and has been a consultant to Pharmacyclics, X4 and BMS; has been a consultant to Janssen and Beigene; and has received research funding from Eli Lilly.
