

Key Points
HHT genotypes are clinically relevant but poorly predicted even by analysis of extensive physical and hematological phenotypic data.
High-throughput DNA sequencing, detailed phenotyping, and statistical and structural modeling contribute to a framework to better prognosticate and treat HHT.

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular dysplasia. Care delivery for HHT patients is impeded by the need for laborious, repeated phenotyping and gaps in knowledge regarding the relationships between causal DNA variants in ENG, ACVRL1, SMAD4 and GDF2, and clinical manifestations. To address this, we analyzed DNA samples from 183 previously uncharacterized, unrelated HHT and suspected HHT cases using the ThromboGenomics high-throughput sequencing platform. We identified 127 rare variants across 168 heterozygous genotypes. Applying modified American College of Medical Genetics and Genomics Guidelines, 106 variants were classified as pathogenic/likely pathogenic and 21 as nonpathogenic (variant of uncertain significance/benign). Unlike the protein products of ACVRL1 and SMAD4, the extracellular ENG amino acids are not strongly conserved. Our inferences of the functional consequences of causal variants in ENG were therefore informed by the crystal structure of endoglin. We then compared the accuracy of predictions of the causal gene blinded to the genetic data using 2 approaches: subjective clinical predictions and statistical predictions based on 8 Human Phenotype Ontology terms. Both approaches had some predictive power, but they were insufficiently accurate to be used clinically, without genetic testing. The distributions of red cell indices differed by causal gene but not sufficiently for clinical use in isolation from genetic data. We conclude that parallel sequencing of the 4 known HHT genes, multidisciplinary team review of variant calls in the context of detailed clinical information, and statistical and structural modeling improve the prognostication and treatment of HHT.
Introduction
Hereditary hemorrhagic telangiectasia (HHT; also known as Osler Weber Rendu syndrome)1 is a vascular dysplasia affecting ∼1 in 6000 people,2-5 with clinical manifestations varying between affected individuals. Inherited as an autosomal dominant6 disease, HHT leads to the development of large visceral arteriovenous malformations (AVMs) and smaller telangiectasias at characteristic mucocutaneous and gastrointestinal sites.7-10 Affected individuals usually need specific management of symptoms, including those resulting from hemorrhage and anemia,11-14 together with screening and multisystemic management programs to prevent future complications and increase life expectancy.1,4,11,14-16 Although clinical manifestations vary by genetic etiology, such programs currently do not distinguish between the major HHT genotypes, potentially exposing some individuals to excessive investigations and treatments and others to insufficient management.
Molecular diagnostics to identify the single allele that is responsible for HHT in any given family offer the clearest route to date for the tailoring of care pathways best suited to the individual, by both ascertaining the presence of HHT, with attendant screening requirements, and subcategorizing according to emerging gene-specific risk profiles.16-26 However, despite earlier recommendations,14 serial data from international surveys indicate that between 2011 and 2018, the proportion of affected families undergoing genetic testing for HHT had only risen from 23.4%27 to 49.4%,28 with threefold differences between countries.28
The majority of HHT patients with a molecular diagnosis have a pathogenic DNA sequence variant in ENG, encoding endoglin (ENG; HHT1),29 or ACVRL1, encoding activin receptor–like kinase (ALK1; HHT2).30 A smaller proportion harbor a pathogenic variant in SMAD4, which can also cause other pathologies, including juvenile polyposis and aortopathy.23-26 Essentially all known HHT pathogenic variants are null alleles. Separate pathophysiological considerations apply to why individual vascular abnormalities then develop at particular sites (eg, specific local triggers in the setting of germ line haploinsufficiency,31 potentially including somatic mosaic loss of the second allele),32 but these considerations are not the focus of the current report.
ENG, ACVRL1, and SMAD4 encode endothelial cell–expressed proteins that transmit or regulate signals by bone morphogenetic protein (BMP)/transforming growth factor-β (TGF-β) superfamily ligands, through heteromeric receptor serine-threonine kinase complexes, to canonical (SMAD-based) and noncanonical (non-SMAD) pathways.33 HHT was initially considered to result from aberrant TGF-β signaling, but newer data implicate the specific ALK1 ligands BMP9 (encoded by GDF2) and BMP10.34 Accordingly, BMP9/10 inhibition recapitulates features of HHT,35 and a direct interaction was detected biochemically between ENG and BMP9/BMP1036 as well as captured by a crystal structure of the endoglin-BMP9 complex determined in 2017.37 GDF2 was considered a candidate gene for HHT because missense substitutions were identified in 3 of 191 unrelated individuals with nosebleeds and skin telangiectasia who did not carry any pathogenic variants in ENG, ACVRL1, or SMAD4.38 Although unreplicated for many years, one GDF2 family with HHT seems to have been identified through the 100 000 Genomes Project.39
Reaching a conclusive molecular diagnosis is complicated by the appreciable proportions of missense variants in the HHT genes to which pathogenicity cannot be assigned without protein or functional studies.40 Early HHT causal gene-phenotype studies focused on the primary structural abnormalities (AVMs/telangiectasia).17-22 We hypothesized that a more systematic approach to HHT molecular diagnostics, including evaluation of both primary and secondary HHT phenotypes, would provide opportunities for the genetic stratification of clinical care. Here, we report the identification and phenotypic integration of 127 variants, of which 64 are novel, across 168 heterozygous genotypes in 147 HHT families.
Methods
The research was approved by national ethics committees, and all human participants provided written informed consent (additional details in supplemental Methods, available on the Blood Web site).
Cohort 1
Cohort 1 (n = 183) comprised unrelated patients attending a single UK institution for management of a clinical diagnosis of known or suspected HHT41,42 where the family’s HHT pathogenic variant was not already known from research43-46 or clinical genotyping. HHT symptoms and features were classified using the Curaçao criteria of nosebleeds, mucocutaneous telangiectasia, visceral involvement, and an affected first-degree relative.47 Using a templated record, 301 Human Phenotype Ontology (HPO) terms,48 along with numerical indices, were recorded from primary notes, radiology systems, and electronic blood records. We also recorded the number and proportion of relatives affected. Overall, cohort 1 comprised 183 probands (113 females [61.7%]).
Cohort 2
Cohort 2 (n = 94) was from the same institution and comprised 94 additional probands (62 females [66.0%]) with a prior clinical and molecular diagnosis, to provide a replicate data set from the same geographical background. Sixty-two had ENG pathogenic variants, 27 ACVRL1, and 5 SMAD4.
Bleeding, red cell, and oxygenation indices (cohorts 1 and 2)
With ethical approval (LREC 00/5792), all available HPO information, bleeding phenotypes, and quantitative red cell and oxygenation indices were collected for both cohorts on the probands and affected family members reviewed in the same clinical service. All red cell and oxygenation indices were measured in the same laboratory: red cell indices on Sysmex XE Series Analysers (Kobe, Japan), and pulse oximetry on Ohmeda Biox 3900s (Boulder, CO). Data collections were performed in 2017 and 2018. Red cell indices were measured by complete blood counts, and oxygen saturation (SaO2 which reduces proportionally to the fraction of right ventricular output passing through pulmonary AVMs) by finger oximetry over 10 minutes standing.49-51 Blinded to genotype, clinical descriptions of patients’ bleeding status were assigned to our institutional 7-point HHT bleeding score which changes dynamically as the patient’s status changes41,52 ; 0 to 4 are based on nosebleeds (0, none; 1, <1 per year; 2, ≤1 per month; 3, several per week; and 4, daily). An extra point is added for states leading to additional iron losses,13 which in this cohort were menorrhagia (n = 4), severe gastrointestinal bleeding (n = 8), blood donation (n = 1), or large-volume near-daily nosebleeds (n = 24). A score of 6 is reserved for patients who are blood transfusion or IV iron dependent at the time, either with daily extreme or torrential nosebleeds or severe gastrointestinal bleeding.13,41,53 Comparative analyses were restricted to the first recorded full data set per patient, irrespective of whether this preceded therapeutic corrections. For binary analyses, severe bleeders were categorized by a bleeding score >4.
Sequencing and bioinformatics
The ThromboGenomics diagnostic HTS platform54 (version 2) was used to identify variants in the exonic fraction of targeted HHT causal genes (ENG, ACVRL1, and SMAD4) and the candidate gene GDF2 using the following transcripts: ENG, LRG_589, NM_000118.3, and NM_001114753.2; ACVRL1, LRG_543 and NM_000020.2; and SMAD4, LRG_318 and NM_005359.5. Relevant intronic regions, untranslated regions, and 1000-bp promoter regions were included. The reads in the demultiplexed paired-end FASTQ files were processed as described previously54 to call single nucleotide variants, indels, and large deletions.
Variant interpretation
Variants were annotated and filtered in an automated fashion using the rules described previously.54 Candidate variants were assessed during multidisciplinary team (MDT) meetings to assign their pathogenicity status for this autosomal dominant disease caused by a single pathogenic null allele in each patient, recognizing the possibility that second variants may influence function sufficiently to modify disease presentation. Where probands carried >1 variant in the HHT genes, evidence for (likely) pathogenic status was sought for all of them. American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) criteria were the primary reference for classifying evidence for pathogenicity in terms of disease causation: for pathogenic impact: very strong [PVS], strong [PS], moderate [PM], and supporting [PP]; for benign impact: standalone [BA], strong [BS], and supporting [BP].55 The letter code is followed by 1 to 7 according to the precise criterion.55 Pathogenicity assignments for some known variants are inconsistent within existing databases (supplemental Figures 1i-ii), and we made minor modifications to improve assignments for this rare disease (supplemental Figure 1iii-vi). The key steps followed by the MDT, discussed further in the supplemental Data, were:
Rare variants met ACMG/AMP criterion PVS1 for pathogenic if they clearly resulted in null alleles (transcript ablation multiexon, panexon, and frameshift insertions/deletions, stop codon gains, and disruption of start or splice site consensus sequences55 ).
Rare inframe indels and missense substitutions were evaluated conservatively, mindful that only ACVRL1 and SMAD4 exhibit appreciable constraint against missense variation. In ExAC, the expected number of missense variants in ACVRL1 was 199.6, but only 132 were observed (z score, 2.34); in SMAD4, 179.6 were expected, but only 65 were observed (z score, 4.18); in ENG, 235.9 were expected, and 223 were observed (z score, 0.41); and in GDF2, 165.7 were expected, and 158 were observed (z score, 0.29).56 In agreement with ExAC, in gnomAD,57 there was also significant constraint for ACVRL1 and SMAD4 but not for ENG and GDF2, showing that ENG and GDF2 have a higher tolerance of missense substitutions than ACVRL1 or SMAD4. Even for SMAD4, amino acid conservation assessed using Consurf58 cautioned us against applying criteria across a full gene. Thus, rare inframe indels and missense substitutions met the rules for likely pathogenic if there was published cellular evidence that they generated null alleles (PS3) and/or they were sited within known functional protein domains of ALK1 (serine-threonine kinase catalytic domain, ligand binding site) or SMAD4 MH2 domain (PM1), and also met a sufficient number of other strong, moderate, or supporting criteria.55
After publication of crystal structures of human endoglin and its complex with BMP9 in June 2017,37 all missense and inframe deletions in ENG were examined, blinded to evolving MDT assignments. Modeling of certain individual variants offered an additional criterion (PM1) to confirm and/or to move a variant of uncertain significance toward likely pathogenic status based on its predicted effect on either the folding of endoglin or the ability of the protein to interact with BMP9.37 Three-dimensional clustering of the variants was also used to identify possible novel functional sites of endoglin.
Any common alleles (allele frequency >0.05 in 1000 Genomes59 ) meeting BA155 were removed by the ThromboGenomics pipeline. Remaining relatively common alleles (allele frequency >0.02 in 1000 Genomes59 ) met BS155 and were designated as likely benign.
Causal gene predictions
Before DNA sequencing, based on the sites and morphology of any telangiectasias, AVMs, and family history, an experienced HHT clinician predicted whether the disease was likely to be HHT, a different inherited vasculopathy masquerading as HHT,60,61 or an isolated AVM (commonly referred to as sporadic1 ). HPO terms for the proband and family were then used to predict which causal gene defect was most likely and provide subjective confidence in these predictions. Key assignment terms were juvenile polyposis (SMAD4 predicted), severe hepatic AVMs or pulmonary arterial hypertension (ACVRL1 predicted), and multiple pulmonary or cerebral AVMs (ENG predicted). In some cases, it was considered possible to exclude 1 gene but not 2; for example, a family with multiple pulmonary or cerebral AVMs and either colorectal polyposis or unusual cancers would have ENG or SMAD4 assigned.
After sequencing, where a pathogenic variant was identified for a cohort 1 DNA, the pre-MDT accuracy of the clinical prediction of the causal gene 1 year earlier was evaluated. Additionally, the positive predictive value, negative predictive value, sensitivity, and specificity of the subjective expert clinician’s call at varying levels of confidence were reviewed for the different molecular subtypes, particularly the more common subtypes of frameshift deletions, splice sites, and nonsense and missense substitutions.
To evaluate if a statistical approach could aid in predicting the causal gene, phenotypes that were thought to discriminate between the 3 established HHT genes were used in a predictive model. For ENG, ACVRL1, and SMAD4, prior beta distributions of frequencies of the discriminatory HPO-coded abnormalities resulting from defects in that gene were assigned according to data from earlier studies,17-26 and using prior clinical observations; for example, the small number of SMAD4 families evaluated previously seemed to resemble ENG more than ACVRL1 in terms of visceral AVMs.23-26 The prevalence of ACVRL1-, ENG-, and SMAD4-related causes of HHT were assumed to be 27%, 67%, and 6%, respectively, based on the genotypes of the families in cohort 2.
The gene indexed by maxkP(g = k|y), which is the gene with the highest posterior probability, was predicted to be the causal gene.
After variant pathogenicity assignment, the correspondence between the gene harboring a likely pathogenic or pathogenic variant as decided by the MDT and the predicted gene (both from manual prediction and statistical prediction) was assessed.
Results
Coverage profile of the ThromboGenomics platform
The mean coverage across the targeted regions of the 4 genes examined (ENG, ACVRL1, SMAD4, and GDF2, comprising 17 878 bp) was on average 832.6× (range, 637.0-1395.0; Figure 1A). The mean fractions of exonic bases covered at 20× and 50× were 0.9999994 and 0.9999404, respectively (Figure 1B). Individual coverage profiles for each gene on the platform showed that virtually all exonic bases of the ThromboGenomics transcripts for these 4 genes are covered sufficiently for sensitive variant calling (supplemental Figure 2), with the profile of GDF2 shown in Figure 1C as a specific example.
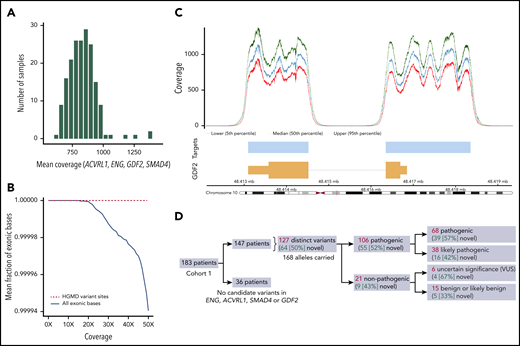
Technical evaluation and output from the HHT panel of the ThromboGenomics platform. (A) Histogram of mean coverage in 183 samples over the targeted regions of the 4 targeted genes (ENG, ACVRL1, SMAD4, and GDF2). (B) The fraction of targeted exonic bases covered at the specified depth (0×-50×) or more, averaged over samples. The solid black line indicates exonic bases and demonstrates that on average, 99.99% of the targeted exonic bases are covered by at least 50 sequencing reads. The dashed red line indicates bases that lie within Human Genome Mutation Database (HGMD) variants and demonstrates that they are all covered by at least 50 sequencing reads. (C) Coverage profile for the GDF2 gene encoding BMP9 on chromosome 10, mapped against the corresponding transcript (orange), which indicates the position and size of the 2 GDF2 exons. The pale blue bars indicate the targeted region, and the 3 traces above indicate the median and 5th and 95th percentile coverage across the locus. Despite the high coverage, no pathogenic variants were identified in the cohort. Equivalent plots for ENG, ACVRL1, and SMAD4 are provided in supplemental Figure 2. (D) Schematic of the classification of the 127 distinct candidate variants identified by the platform. VUS, variant of uncertain significance.
Technical evaluation and output from the HHT panel of the ThromboGenomics platform. (A) Histogram of mean coverage in 183 samples over the targeted regions of the 4 targeted genes (ENG, ACVRL1, SMAD4, and GDF2). (B) The fraction of targeted exonic bases covered at the specified depth (0×-50×) or more, averaged over samples. The solid black line indicates exonic bases and demonstrates that on average, 99.99% of the targeted exonic bases are covered by at least 50 sequencing reads. The dashed red line indicates bases that lie within Human Genome Mutation Database (HGMD) variants and demonstrates that they are all covered by at least 50 sequencing reads. (C) Coverage profile for the GDF2 gene encoding BMP9 on chromosome 10, mapped against the corresponding transcript (orange), which indicates the position and size of the 2 GDF2 exons. The pale blue bars indicate the targeted region, and the 3 traces above indicate the median and 5th and 95th percentile coverage across the locus. Despite the high coverage, no pathogenic variants were identified in the cohort. Equivalent plots for ENG, ACVRL1, and SMAD4 are provided in supplemental Figure 2. (D) Schematic of the classification of the 127 distinct candidate variants identified by the platform. VUS, variant of uncertain significance.
Variant calling
Automated variant filtering yielded 168 heterozygous candidate variant calls within the 4 examined genes in 147 of the 183 patients in cohort 1. The remaining 36 patients had no candidate variants identified. Among the 168 candidate variant calls, 127 were unique across the cohort, 64 (50.4%) of which had not been previously identified in HHT cases. The full list of candidate variants is provided in supplemental Table 1.
Variants were brought to MDT meetings for pathogenicity assignment. Among the 127 unique variants, 106 were labeled as pathogenic or likely pathogenic, providing a molecular diagnosis in 140 (74.9%) of 183 patients (Figure 1D). The 68 rare variants assigned as pathogenic and 38 rare variants as likely pathogenic spanned genomic structural variants, indels, consensus splice variants, and single nucleotide variants affecting amino acid residues within critical functional domains of the ALK1 or SMAD4 proteins, noting these were not always mutually exclusive (supplemental Table 2; supplemental Figure 3).
In contrast to ALK1 and SMAD4 proteins, extracellular ENG amino acids displayed highly variable normalized conservation scores (Figure 2), implying that domain-based, secondary structural predictions were unlikely to be feasible. The 3-dimensional mapping of likely pathogenic ENG missense and inframe indels to the crystallographic model of extracellular endoglin37 suggested that previously unsuspected regions of the protein are important for its function (Figure 3), thus implicating noncontiguous amino acids as being of particular functional importance. Specifically, a hotspot for pathogenic variants was identified in α-helix 2 and β-strand 16 of orphan domain 1, adjacent to the region that directly contacts BMP9 (Figure 3Ai-ii).
![Normalized amino acid conservation scores across ENG, ALK1, and SMAD4. The degree of evolutionary conservation of each amino acid in the human protein sequences of ENG (NM_0011147, 658 amino acids), ACVRL1 (NM_000020.2, 503 amino acids), and SMAD4 (NM_005359.5, 552 amino acids), plotted against the respective amino acid position. The conservation, reflecting the retention of macromolecular function, was plotted as normalized conservation scores and 95% confidence intervals (CIs) obtained using ConSurf.58 Lower scores indicate greater conservation. In all 6 plots, the selected amino acids are plotted in red, and all other amino acids are plotted in black. (A) Amino acid sites of pathogenic or likely pathogenic HHT missense substitutions from current cohort 1 (n = 18), cohort 2 (n = 15), and the 2018 HHT Mutation Database40 (n = 64) are plotted in red, and all other amino acids are plotted in black. Amino acids in which pathogenic or likely pathogenic variants were located were more conserved than amino acids with nonpathogenic variants (ENG mean difference, −0.52 [95% CI, −0.81 to −0.24; P = .00072]; ACVRL1, −0.77 [95% CI, −0.94 to −0.61; P = 6.6x10−15]; SMAD4, −0.80 [95% CI, −0.89 to −0.72; P = 3.9 × 10−61]). Notably, however, not all pathogenic or likely pathogenic variants were at conserved sites, and for endoglin, the normalized conservation scores and CIs were highly variable in regions other than the transmembrane domain (near amino acid 600) and the C terminal cytoplasmic tail (amino acids 635-658). (B) Amino acid sites of likely benign missense substitutions in the gnomAD database57 plotted in red vs all other amino acids, plotted in black. Amino acids in which benign variants were sited were less conserved than other amino acids (ENG mean difference, 0.34 [95% CI, 0.19-0.50; P = 1.6 × 10−5]; ACVRL1, 0.48 [95% CI, 0.30-0.66; P = 3.2 × 10−7]; SMAD4, 0.54 [95% CI, 0.32-0.77; P = 3.0 × 10−6]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/17/10.1182_blood.2019004560/1/m_bloodbld2019004560f2.png?Expires=1769095335&Signature=Z~OcSmjcZO04FrVksp0w1IBZLQx9S6BwIE~RQaX~WhQEMJugYJdOyHy1R~HHW1JkoFTJU3LzsZRbGrCyBvGL-dNu5611bpLtJ1L6xswiaShgbZwm47KhjTaSdaw4-3sA-pQCLZ~0ihQwGNY95GU7RwC8YYh1mFqeo5ptJqFpC-dBvHS3opKxy99~84doClcBCLv9ID-798ztIp02Rwy-mfDV2Y5J-pv~h-9t94i1r9kCZ437273o491gRuCZaBjO9kNdMraZTdssJxsGEcWZeqMKL0DehwSM6go2czYkpeNBTJW0a33Q2HIEuCdcMRlAX9tiaVkzidwSZL8Lw134Iw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Normalized amino acid conservation scores across ENG, ALK1, and SMAD4. The degree of evolutionary conservation of each amino acid in the human protein sequences of ENG (NM_0011147, 658 amino acids), ACVRL1 (NM_000020.2, 503 amino acids), and SMAD4 (NM_005359.5, 552 amino acids), plotted against the respective amino acid position. The conservation, reflecting the retention of macromolecular function, was plotted as normalized conservation scores and 95% confidence intervals (CIs) obtained using ConSurf.58 Lower scores indicate greater conservation. In all 6 plots, the selected amino acids are plotted in red, and all other amino acids are plotted in black. (A) Amino acid sites of pathogenic or likely pathogenic HHT missense substitutions from current cohort 1 (n = 18), cohort 2 (n = 15), and the 2018 HHT Mutation Database40 (n = 64) are plotted in red, and all other amino acids are plotted in black. Amino acids in which pathogenic or likely pathogenic variants were located were more conserved than amino acids with nonpathogenic variants (ENG mean difference, −0.52 [95% CI, −0.81 to −0.24; P = .00072]; ACVRL1, −0.77 [95% CI, −0.94 to −0.61; P = 6.6x10−15]; SMAD4, −0.80 [95% CI, −0.89 to −0.72; P = 3.9 × 10−61]). Notably, however, not all pathogenic or likely pathogenic variants were at conserved sites, and for endoglin, the normalized conservation scores and CIs were highly variable in regions other than the transmembrane domain (near amino acid 600) and the C terminal cytoplasmic tail (amino acids 635-658). (B) Amino acid sites of likely benign missense substitutions in the gnomAD database57 plotted in red vs all other amino acids, plotted in black. Amino acids in which benign variants were sited were less conserved than other amino acids (ENG mean difference, 0.34 [95% CI, 0.19-0.50; P = 1.6 × 10−5]; ACVRL1, 0.48 [95% CI, 0.30-0.66; P = 3.2 × 10−7]; SMAD4, 0.54 [95% CI, 0.32-0.77; P = 3.0 × 10−6]).
Normalized amino acid conservation scores across ENG, ALK1, and SMAD4. The degree of evolutionary conservation of each amino acid in the human protein sequences of ENG (NM_0011147, 658 amino acids), ACVRL1 (NM_000020.2, 503 amino acids), and SMAD4 (NM_005359.5, 552 amino acids), plotted against the respective amino acid position. The conservation, reflecting the retention of macromolecular function, was plotted as normalized conservation scores and 95% confidence intervals (CIs) obtained using ConSurf.58 Lower scores indicate greater conservation. In all 6 plots, the selected amino acids are plotted in red, and all other amino acids are plotted in black. (A) Amino acid sites of pathogenic or likely pathogenic HHT missense substitutions from current cohort 1 (n = 18), cohort 2 (n = 15), and the 2018 HHT Mutation Database40 (n = 64) are plotted in red, and all other amino acids are plotted in black. Amino acids in which pathogenic or likely pathogenic variants were located were more conserved than amino acids with nonpathogenic variants (ENG mean difference, −0.52 [95% CI, −0.81 to −0.24; P = .00072]; ACVRL1, −0.77 [95% CI, −0.94 to −0.61; P = 6.6x10−15]; SMAD4, −0.80 [95% CI, −0.89 to −0.72; P = 3.9 × 10−61]). Notably, however, not all pathogenic or likely pathogenic variants were at conserved sites, and for endoglin, the normalized conservation scores and CIs were highly variable in regions other than the transmembrane domain (near amino acid 600) and the C terminal cytoplasmic tail (amino acids 635-658). (B) Amino acid sites of likely benign missense substitutions in the gnomAD database57 plotted in red vs all other amino acids, plotted in black. Amino acids in which benign variants were sited were less conserved than other amino acids (ENG mean difference, 0.34 [95% CI, 0.19-0.50; P = 1.6 × 10−5]; ACVRL1, 0.48 [95% CI, 0.30-0.66; P = 3.2 × 10−7]; SMAD4, 0.54 [95% CI, 0.32-0.77; P = 3.0 × 10−6]).
![Molecular characterization of sequence variants. (A) Mapping of ENG missense substitutions in cohorts 1 and 2 onto the crystal structures of ENG and its complex with BMP9. Proteins are shown in cartoon representation (BMP9, yellow), with specific ENG amino acids depicted as sticks and carbon atoms colored dark magenta and N-glycosylation site N307 colored cyan. (i) The relative position of 6 pathogenic or likely pathogenic variants described in the present report (Cys[C]207Tyr, Leu[L]300Pro, Leu[L]299Arg, Ile[I]220Asn, Leu[L]221Gln, and Asn[N]307Leu) defines a hotspot for pathogenic or likely pathogenic missense variants, which includes the α-helix 2 of the ENG OR1 domain, the C-terminal end of which lies close to the BMP9 binding site.37 (ii) A second hotspot for pathogenic or likely pathogenic variants (Lys[K]216Glu and Glu[E]217delinsGluAla) affects residues at the N-terminal end of OR1 β-strand 16, including Lys216, which connects the C-terminal end of α-helix 2 to Gln270 at the ENG/BMP9 interface via hydrogen bond interactions. This view, which depicts amino acid contacts as observed in the high-resolution structure of ENG OR,37 also shows the location of the Cys(c)207Tyr and Ile(I)220Asn mutations from a different perspective. (iii) The duplication of Leu(L)170 affects residues in the core of ENG OR2, where Leu170 is involved in a number of hydrophobic interactions. The duplication most likely affects the folding of ENG, rather than directly affecting its function; the insertion could either disrupt the register of the N-terminal part of OR2 β-strand 12 or, more likely, cause an additional residue to be accommodated in the loop that follows the same β-strand. In the latter case, the extra Leu would take the place of Arg(R)171, disrupting a hydrogen bond with Glu(E)195 as well as a stacking interaction with Arg(R)192. Moreover, by shifting Arg(R)171 to take the place of Leu(L)172, the duplication would replace a hydrophobic residue with a charged residue at the bottom of the hydrophobic core of the OR2 β-sandwich. (iv) Three variants that are benign in terms of HHT pathogenesis (Gly413Asp, Asp446Gly, and Arg571His) affect residues that are all exposed on the same face of the ENG ZP module.37 The 3 variants were independently assigned as benign without reference to the tertiary structure because of their presence in the same HHT DNA as an ENG nonsense (stop), splice, or frameshift variant, respectively. (B) Bar plot of the number of pathogenic or likely pathogenic variants in the HHT Mutation Database (DB) and in cohort 1, broken down by sequence ontology (SO) term in ENG, ACVRL1, and SMAD4. The upper bars give the number in the HHT Mutation DB in 201840 (620 variants in total); the lower bars give the number in cohort 1, with novel variants highlighted in dark blue (106 variants in total). TG, ThromboGenomics; UTR, untranslated region.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/17/10.1182_blood.2019004560/1/m_bloodbld2019004560f3.png?Expires=1769095335&Signature=aq0fpN1W9pCcDzfIDm~pAJu33HNtJ~5MLByYaRCXu4P4YyowJRU~gaX5wMwvM0uyxFC2KXCVdUJu50F4JvdX5kPCm48hHU0ULEavTcpxGwGEMSJALtwu1Atj0mFF-IoxvCy0-lcIADo8BukbvO04WNUt6yFIFS4AFrX4yda9F-f8dhjgCdeh2TyZTRrjW5TjrdHwValmDFRqJ0aOr6TgkS3ET7QfxnH9PJQFQsaDHFnY3eqImVwX~1eWndllZbtAQk50yl6INfp8QumonCT3P1RkGXnzIT3VP6O3sAD08lagtsT7PNh6cSZHMJ1qSjts0ZHPVU1qx9zhIRSWAIn2iQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Molecular characterization of sequence variants. (A) Mapping of ENG missense substitutions in cohorts 1 and 2 onto the crystal structures of ENG and its complex with BMP9. Proteins are shown in cartoon representation (BMP9, yellow), with specific ENG amino acids depicted as sticks and carbon atoms colored dark magenta and N-glycosylation site N307 colored cyan. (i) The relative position of 6 pathogenic or likely pathogenic variants described in the present report (Cys[C]207Tyr, Leu[L]300Pro, Leu[L]299Arg, Ile[I]220Asn, Leu[L]221Gln, and Asn[N]307Leu) defines a hotspot for pathogenic or likely pathogenic missense variants, which includes the α-helix 2 of the ENG OR1 domain, the C-terminal end of which lies close to the BMP9 binding site.37 (ii) A second hotspot for pathogenic or likely pathogenic variants (Lys[K]216Glu and Glu[E]217delinsGluAla) affects residues at the N-terminal end of OR1 β-strand 16, including Lys216, which connects the C-terminal end of α-helix 2 to Gln270 at the ENG/BMP9 interface via hydrogen bond interactions. This view, which depicts amino acid contacts as observed in the high-resolution structure of ENG OR,37 also shows the location of the Cys(c)207Tyr and Ile(I)220Asn mutations from a different perspective. (iii) The duplication of Leu(L)170 affects residues in the core of ENG OR2, where Leu170 is involved in a number of hydrophobic interactions. The duplication most likely affects the folding of ENG, rather than directly affecting its function; the insertion could either disrupt the register of the N-terminal part of OR2 β-strand 12 or, more likely, cause an additional residue to be accommodated in the loop that follows the same β-strand. In the latter case, the extra Leu would take the place of Arg(R)171, disrupting a hydrogen bond with Glu(E)195 as well as a stacking interaction with Arg(R)192. Moreover, by shifting Arg(R)171 to take the place of Leu(L)172, the duplication would replace a hydrophobic residue with a charged residue at the bottom of the hydrophobic core of the OR2 β-sandwich. (iv) Three variants that are benign in terms of HHT pathogenesis (Gly413Asp, Asp446Gly, and Arg571His) affect residues that are all exposed on the same face of the ENG ZP module.37 The 3 variants were independently assigned as benign without reference to the tertiary structure because of their presence in the same HHT DNA as an ENG nonsense (stop), splice, or frameshift variant, respectively. (B) Bar plot of the number of pathogenic or likely pathogenic variants in the HHT Mutation Database (DB) and in cohort 1, broken down by sequence ontology (SO) term in ENG, ACVRL1, and SMAD4. The upper bars give the number in the HHT Mutation DB in 201840 (620 variants in total); the lower bars give the number in cohort 1, with novel variants highlighted in dark blue (106 variants in total). TG, ThromboGenomics; UTR, untranslated region.
Molecular characterization of sequence variants. (A) Mapping of ENG missense substitutions in cohorts 1 and 2 onto the crystal structures of ENG and its complex with BMP9. Proteins are shown in cartoon representation (BMP9, yellow), with specific ENG amino acids depicted as sticks and carbon atoms colored dark magenta and N-glycosylation site N307 colored cyan. (i) The relative position of 6 pathogenic or likely pathogenic variants described in the present report (Cys[C]207Tyr, Leu[L]300Pro, Leu[L]299Arg, Ile[I]220Asn, Leu[L]221Gln, and Asn[N]307Leu) defines a hotspot for pathogenic or likely pathogenic missense variants, which includes the α-helix 2 of the ENG OR1 domain, the C-terminal end of which lies close to the BMP9 binding site.37 (ii) A second hotspot for pathogenic or likely pathogenic variants (Lys[K]216Glu and Glu[E]217delinsGluAla) affects residues at the N-terminal end of OR1 β-strand 16, including Lys216, which connects the C-terminal end of α-helix 2 to Gln270 at the ENG/BMP9 interface via hydrogen bond interactions. This view, which depicts amino acid contacts as observed in the high-resolution structure of ENG OR,37 also shows the location of the Cys(c)207Tyr and Ile(I)220Asn mutations from a different perspective. (iii) The duplication of Leu(L)170 affects residues in the core of ENG OR2, where Leu170 is involved in a number of hydrophobic interactions. The duplication most likely affects the folding of ENG, rather than directly affecting its function; the insertion could either disrupt the register of the N-terminal part of OR2 β-strand 12 or, more likely, cause an additional residue to be accommodated in the loop that follows the same β-strand. In the latter case, the extra Leu would take the place of Arg(R)171, disrupting a hydrogen bond with Glu(E)195 as well as a stacking interaction with Arg(R)192. Moreover, by shifting Arg(R)171 to take the place of Leu(L)172, the duplication would replace a hydrophobic residue with a charged residue at the bottom of the hydrophobic core of the OR2 β-sandwich. (iv) Three variants that are benign in terms of HHT pathogenesis (Gly413Asp, Asp446Gly, and Arg571His) affect residues that are all exposed on the same face of the ENG ZP module.37 The 3 variants were independently assigned as benign without reference to the tertiary structure because of their presence in the same HHT DNA as an ENG nonsense (stop), splice, or frameshift variant, respectively. (B) Bar plot of the number of pathogenic or likely pathogenic variants in the HHT Mutation Database (DB) and in cohort 1, broken down by sequence ontology (SO) term in ENG, ACVRL1, and SMAD4. The upper bars give the number in the HHT Mutation DB in 201840 (620 variants in total); the lower bars give the number in cohort 1, with novel variants highlighted in dark blue (106 variants in total). TG, ThromboGenomics; UTR, untranslated region.
Of the 147 patients in whom a candidate variant was identified, 5 carried missense variants of uncertain significance. Twenty (13.6%) had 2 or more variants within the 4 analyzed genes (supplemental Table 3). In 19 of these 20 patients, 1 of the variants was a high-impact allele in ACVRL1, ENG, or SMAD4 that explained the HHT phenotype, and the other variants were relatively common (allele frequency >0.0259 ) missense variants and for this reason labeled as benign. Focusing on ENG missense variants designated likely pathogenic (n = 24) and benign (n = 14), it was notable that only 1 of the 14 benign variants (7.1%) was located in a potentially critical region of the tertiary structure, compared with 18 (75.0%) of 24 of the pathogenic missense substitutions (supplemental Table 2C). For 3 benign variants, the near-linear 3-dimensional arrangement (Figure 3Aiv) suggested that amino acid substitutions in 1 of the faces of the ZP module of endoglin do not generate the null alleles required for the HHT phenotype.
As noted, at least 1 variant was identified within the 4 genes in 147 (80.3%) of the 183 previously uncharacterized, unrelated HHT patients with definite or suspected HHT, but not all of these variants were pathogenic. When restricting to the 150 unrelated individuals in cohort 1 meeting ≥3 Curaçao criteria (ie, definite clinical diagnosis of HHT),47,63 125 (83.3%) had a pathogenic variant, providing a clinical diagnostic yield of 125 (83.3%) of 150.
Comparison with previously published HHT cohorts
Pathogenic variants in cohorts 1 and 2 were compared with HHT Mutation Database40 entries on 24 November 2018. This version differed from a previous version from 2012 because of the recent institution of stringent ACMG/AMP criteria55 and consequently contained fewer pathogenic missense substitutions (supplemental Figure 1i). The distribution of the molecular types of variants in cohort 1 was similar to that reported for the 2018 HHT Mutation Database. This correspondence was greatest for the most common causal gene ENG (Figure 3B), despite 38 (52.1%) of the 74 cohort 1 ENG pathogenic variants being newly described.
Four candidate variants were identified in GDF2 in 4 individuals; 1 was a synonymous splice site–adjacent variant in an individual with an ENG missense variant; 3 were missense variants (including 1 previously reported as being pathogenic [GDF2 c.997C>T (p.Arg333Trp)]38 ), but were identified in individuals with a rare likely pathogenic ENG variant and were therefore assigned benign status (supplemental Table 4).
Predicted and observed phenotypes
Of the 301 distinct HPO terms assigned to cases in the current cohort, 8 were predicted a priori to differ in prevalence between patients of different HHT genotypes.17-26 Figure 4A compares the predicted frequency of these phenotypes in each genotype based on existing literature17-26 (triangles) and the observed frequencies across cohorts 1 and 2 (circles). Generally, these concurred, with notable exceptions where screening of asymptomatic patients was not performed in the cohort. Particularly, observed frequencies were lower than predicted for colorectal polyposis in SMAD4 and hepatic AVMs in ACVRL1 (Figure 4A).
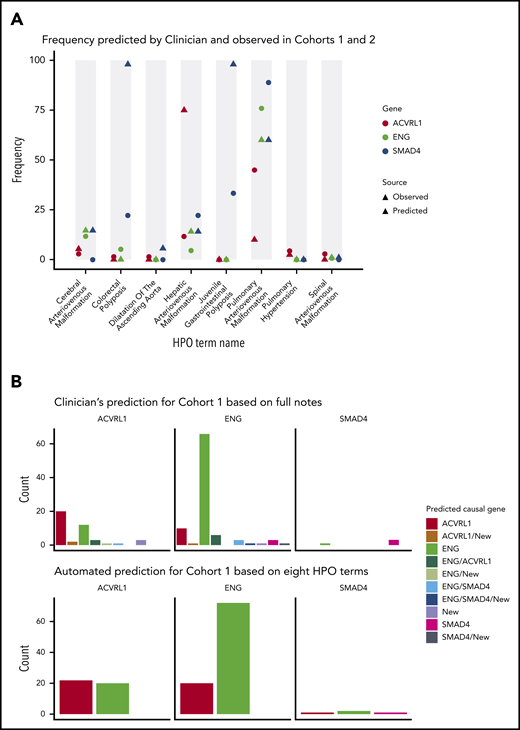
Phenotypic prediction accuracy for samples with pathogenic variants. (A) The predicted (triangle) and observed (circle) frequencies of 8 a priori discriminatory HPO terms in cohorts 1 and 2. (B) Predicted causal gene displayed for each of the observed HHT genotypes in cohort 1, for clinician prediction using all HPO terms (upper panel; additional details in supplemental Table 5), and automated prediction through Bayesian modeling of the 8 discriminatory HPO terms (lower panel; additional details in supplemental Methods, supplemental Figure 4, and supplemental Table 6).
Phenotypic prediction accuracy for samples with pathogenic variants. (A) The predicted (triangle) and observed (circle) frequencies of 8 a priori discriminatory HPO terms in cohorts 1 and 2. (B) Predicted causal gene displayed for each of the observed HHT genotypes in cohort 1, for clinician prediction using all HPO terms (upper panel; additional details in supplemental Table 5), and automated prediction through Bayesian modeling of the 8 discriminatory HPO terms (lower panel; additional details in supplemental Methods, supplemental Figure 4, and supplemental Table 6).
Before genotyping, the suspected causal genes of the probands were assigned based on the distribution of clinical phenotypes within their families. The clinician’s prior suspected genes for probands subsequently demonstrated to have ACVRL1, ENG, or SMAD4 causal variants are illustrated in Figure 4B (upper panel). Overall, positive predictive values for the clinician’s ENG calls were consistently higher than for ACVRL1 (≥85.0% vs ≤70%; all P values <.001), and negative predictive values for SMAD4 were ≥99% (supplemental Table 5). The lower panel shows the distribution of the suspected causal genes from statistical predictions using the 8 HPO terms predicted to be discriminatory. The automated predictions provided very similar predictive accuracy compared with predictions by an expert clinician, particularly for ENG (Figure 4B), although SMAD4 phenotypes were less well predicted by the statistical model.
Erythrocytic indices
To test whether quantitative measurements might enhance predictions of the causal gene, red cell indices were examined, because anemia is both a challenging problem in HHT8,13 and a driver of higher cardiac outputs, flow, and potentially AVM enlargement.64-66 As expected, across cohorts 1 and 2, a subset of HHT cases demonstrated extremely low hematocrit or hemoglobin relative to 50 000 healthy controls (blood donors from the INTERVAL study67 ; Figure 5A). Notably, some HHT cases had elevated red blood cell counts (Figure 5A). However, half or more of HHT patients had red blood cell indices within the central 90% of values in the healthy controls (Figure 5A).
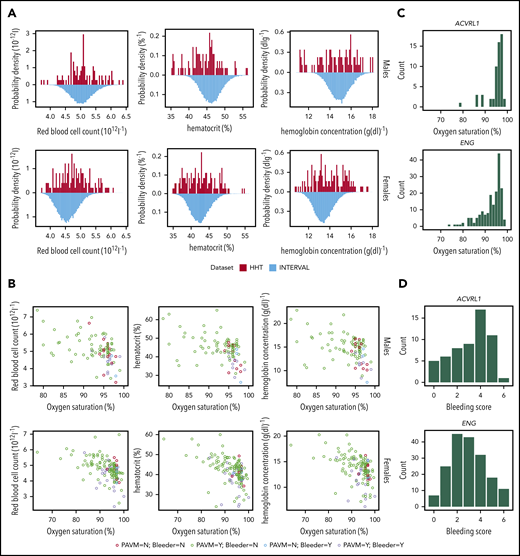
Quantitative red cell traits. (A) Distributions of quantitative red cell traits in HHT and control populations: total red blood cell count (left), hematocrit (center), and hemoglobin (right) plotted for the HHT patients (1 measurement per patient, proband, and affected family members from cohorts 1 and 2) above the respective INTERVAL population distribution from 50 000 blood donors (1 result per donor).67 Upper panel, males; lower panel, females. Although the median values are similar, it should be noted that HHT cases had a higher proportion of extreme red cell values (both high and low) relative to healthy controls; for red cell count, hematocrit, and hemoglobin, respectively, the proportion of HHT patients within the fifth to 95th sex-stratified percentiles of the INTERVAL ranges were only 66%, 61%, and 50% for males and 64%, 55%, and 48% for females, respectively (all P values <.0001). (B) Relationships with bleeding and hypoxemia in HHT cohort. Patients with more severe blood losses (bleeders) were defined by a bleeding score ≥4 and subcategorized by the presence (purple symbols) or absence (blue symbols) of pulmonary AVMs (PAVMs), which impair gas exchange, resulting in lower arterial partial pressure of oxygen and hence lower SaO2. Patients with lower bleeding scores were also categorized by the presence (green) and absence (red) of PAVMs. The graphs (upper panel, males; lower panel, females) plot total red blood cell count (left), hematocrit (center), and hemoglobin (right) against same-day SaO2 measured by finger oximetry for 10 minutes standing using 1 measurement per patient (proband and affected family members). Note that in all 6 analyses, the patients with greater bleeding (red and purple) tended to have lower red blood cell indices (P < .0001 in all cases), and there was a superimposed anticorrelation between the red cell indices and SaO2 (P < .0001 in all cases). (C) SaO2 in HHT patients. Histograms of SaO2 in ACVRL1 and ENG cases. (D) Bleeding score in HHT patients. Histograms of bleeding scores in ACVRL1 and ENG cases.
Quantitative red cell traits. (A) Distributions of quantitative red cell traits in HHT and control populations: total red blood cell count (left), hematocrit (center), and hemoglobin (right) plotted for the HHT patients (1 measurement per patient, proband, and affected family members from cohorts 1 and 2) above the respective INTERVAL population distribution from 50 000 blood donors (1 result per donor).67 Upper panel, males; lower panel, females. Although the median values are similar, it should be noted that HHT cases had a higher proportion of extreme red cell values (both high and low) relative to healthy controls; for red cell count, hematocrit, and hemoglobin, respectively, the proportion of HHT patients within the fifth to 95th sex-stratified percentiles of the INTERVAL ranges were only 66%, 61%, and 50% for males and 64%, 55%, and 48% for females, respectively (all P values <.0001). (B) Relationships with bleeding and hypoxemia in HHT cohort. Patients with more severe blood losses (bleeders) were defined by a bleeding score ≥4 and subcategorized by the presence (purple symbols) or absence (blue symbols) of pulmonary AVMs (PAVMs), which impair gas exchange, resulting in lower arterial partial pressure of oxygen and hence lower SaO2. Patients with lower bleeding scores were also categorized by the presence (green) and absence (red) of PAVMs. The graphs (upper panel, males; lower panel, females) plot total red blood cell count (left), hematocrit (center), and hemoglobin (right) against same-day SaO2 measured by finger oximetry for 10 minutes standing using 1 measurement per patient (proband and affected family members). Note that in all 6 analyses, the patients with greater bleeding (red and purple) tended to have lower red blood cell indices (P < .0001 in all cases), and there was a superimposed anticorrelation between the red cell indices and SaO2 (P < .0001 in all cases). (C) SaO2 in HHT patients. Histograms of SaO2 in ACVRL1 and ENG cases. (D) Bleeding score in HHT patients. Histograms of bleeding scores in ACVRL1 and ENG cases.
Male and female HHT patients with more severe bleeding (bleeding score >4) tended to have lower red cell counts, hematocrit and hemoglobin values than individuals without severe bleeding (Mann Whitney P < .0001 in all cases; Figure 5B). This is in keeping with bleeding leading to iron-restricted erythropoiesis. However, there was a significant anticorrelation between these 3 red cell indices and SaO2 (P < .0001; Figure 5B) because pulmonary AVM-induced hypoxemia leads to compensatory secondary erythrocytosis.68,69
Figure 5C compares these quantitative HHT phenotypes between HHT genotypes. There was a suggestive trend for bleeding score in ACVRL1 cases to be higher than in ENG cases and clear evidence that pulmonary AVM-induced hypoxemia was more severe in ENG patients (Figure 5C; P < .001). However, there was no statistically significant difference in red cell traits between ENG and ACVRL1 cases with or without adjustment for sex, SaO2, and iron (data not shown). Any associations between the HHT causal gene and red cell traits were mediated by bleeding severity and SaO2 and thus red cell indices did not provide opportunities for improving our predictive model.
Discussion
In this study, we applied standardized phenotyping, predictive modeling, and interpretation of high-throughput sequencing at several hundred–fold depth. By identifying 106 pathogenic variants, of which 55 (52%) were newly described, this study provided a confirmed molecular diagnosis for 140 HHT families and focused attention on the HHT genes and functional deductions from HHT causal variants.
All HHT pathogenic variants were heterozygous changes in the established HHT genes ENG, ACVRL1, and SMAD4, but not all rare variants identified in those genes are pathogenic. Substantial work may be required to unravel which inframe indels and missense, nonconsensus splice site, and intronic variants affect protein function sufficiently to produce null, HHT-causing alleles. Hypomorphic, nonnull variants may affect protein function and/or modify disease severity, but based on our current understanding, these would not be the causal alleles segregating in the family to cause this autosomal dominant disease.
The current study demonstrates that clinician predictions alone cannot indicate the HHT causal gene with sufficient confidence, and formal sequencing analysis of the 4 genes included in this study is indicated. Although negative results would not currently change clinical management of the patient, a positive result, which is more likely using the presented methods (supplemental Figure 1vi), allows distinction from other vasculopathies with lesser or different requirements for visceral surveillance60,61 and leads to additional changes in HHT patient management; for example, SMAD4 patients need to enter endoscopic and echocardiographic surveillance programs,24-26 and the presence of a causal ACVRL1 variant increases the perceived risk of hepatic AVMs that may lead to changes in care pathways; there are also early suggestions that drug adverse event profiles may differ according to the causal HHT gene, thus influencing prescription choices.16 Differing natural history data according to the causal gene may permit tailored risk-benefit weightings for other management elements, such as repeat imaging/radiation exposure70,71 or HHT interventions with complications in addition to efficacy.71-75 We speculate that the higher clinician accuracy in predicting ENG nonsense substitutions compared with frameshift indels and splice site variants (supplemental Table 5) may reflect more complex and functional consequences in the latter 2, although this needs to be examined in additional studies. Our data confirm that the value of HHT gene testing extends beyond predictive testing of relatives, and we encourage funding within mainstream medicine.
The proportion of unsolved cases (36 [19.6%] of 183 patients) demonstrates that an inconclusive HHT gene test does not exclude a diagnosis of HHT, unless this has excluded the known HHT pathogenic variant in the family. Further molecular testing is then indicated in due course, while the patient continues to be managed for HHT. Statistical modeling from readily assessable HPO terms offers predictive value, and the approach is adaptable to particular characteristics of the population to account for local screening/referral patterns and detection rates. Future statistical models of the causal gene could incorporate quantitative traits that expose substantial processes influencing HHT phenotypes (Figure 5). As for other recently published clinical measurements (spirometry,76 haptoglobin/hemolysis41 ), these allow evaluation of HHT pathophysiology beyond the championed cellular processes of angiogenesis, vascular repair, and endothelial-mural cell communications.77-82 We cannot rule out hypomorphic alleles having pathogenic roles in specific settings, but we suggest variant labeling as a potential disease modifier should also take into account the wider clinical phenotypic influences from large population-based studies. The latter include not only bleeding phenotypes and SaO2 (as in the current report), but also other hemodynamic factors, such as systemic vascular resistance, cardiac output, and cardiac function.
Our results lead us to propose the following systematic approaches to HHT care, incorporating:
Molecular confirmation/exclusion of HHT status to support preventative strategies while limiting unnecessary investigations; parallel panel-based, high-depth sequencing across HHT causal and candidate genes seems optimal, enabling non-HHT causal variants to be cataloged for clinical and research use.
A systematic approach to the application of ACMG Guidelines for a standardization of variant interpretation and recording of all variant classifications by date.
Systematic capture of clinical measures that influence the HHT phenotype; at a minimum, we suggest these include a current broad bleeding score, and SaO2, which is not only a biomarker for severity and risk of pulmonary AVMs, but also a major influencer of hemodynamic and hematological phenotypes in HHT.
We conclude that high-throughput, high-depth sequencing platforms of HHT causal genes, statistical and structural modeling of heterozygous, potentially null alleles, and standardized phenotyping are the methods of choice in the 21st century.
The variants are available in ClinVar (under "ThromboGenomics_HHT") and the HHT Mutation Database.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Hammersmith Hospital, Imperial College Healthcare National Health Service (NHS) Trust for patient recruitment to Imperial College–sponsored programs, ethically approved by the National Research Ethics Committee East of Scotland Multicentre Research Ethics Committee (MREC/98/0/42, LREC 99/5637M, and 07/MRE00/19). The authors also gratefully acknowledge colleagues and staff at Hammersmith Hospital and Imperial College Healthcare NHS Trust for assistance with patient phenotyping as part of standard NHS care, as well as the patients for their willing cooperation in these studies. The authors acknowledge the Cambridge National Institute for Health Research (NIHR) BioResource Centre, NIHR, and National Health Service Blood and Transplant for their contribution to the ThromboGenomics platform.
This work was supported by the NIHR BioResource Scheme, which funded and developed the ThromboGenomics platform (NIHR RG65966). C.L.S.'s patient-facing and molecular research was cofunded by the NIHR Imperial Biomedical Research Centre, the Margaret Straker Memorial Trust, the Averil Macdonald Memorial Fund, the Wellcome Trust (TF/037257, AF/053286), and the British Heart Foundation (PG/2000067, FS/04/089, PG/09/041/27515). I.S., K.M., C.J.P., and E.T. are supported by NIHR BioResource–Rare Diseases (NIHR RG65966). K.D. is supported as an NHS Higher Specialist Scientist Training trainee by Health Education England and NIHR BioResource–Rare Diseases (NIHR RG65966). W.H.O. is supported by RBAG/181 NIHR BioResource–Rare Diseases (NIHR RG65966), the British Heart Foundation (RBAG/245, RBAG/208, RBAG/226), the European Commission (RBAG/344), the Medical Research Council (RBAG/285, RBAG/295), NHS Blood and Transplant (RBAG/142), and the Wellcome Trust (RBAG/342). L.J. is supported by the Swedish Research Council (2016-03999), the Center for Innovative Medicine (2-537/2014), and Karolinska Institutet research foundations (2018-02166). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
Authorship
Contribution: C.L.S. obtained ethical approvals, performed all clinical studies, consented families, extracted DNA, predicted causal genes, led the multidisciplinary team (MDT), defined and applied the ACMG-based algorithm, wrote the first manuscript draft and data supplement, developed the text and figures with E.T. and L.J., and revised and approved the final manuscript; I.S. contributed to ThromboGenomics platform design, managed ThromboGenomics, processed samples, and reviewed and approved the final manuscript; K.D. managed ThromboGenomics and reviewed and approved the final manuscript; K.M. contributed to ThromboGenomics platform design, performed gene and transcript curation, submitted variants to the public repository, and reviewed and approved the final manuscript; Z.C.F. extracted DNA, contributed to data discussions, and reviewed and approved the final manuscript; M.E.B.-H. participated in the MDT, contributed to data discussions, and approved the final manuscript; A.S. performed database comparisons presented in supplemental Figure 1ii and approved the final manuscript; J.B. recruited study participants, contributed to data discussions, and reviewed and approved the final manuscript; D.P. extracted DNA, contributed to data discussions, and reviewed and approved the final manuscript; L.K. assisted with study phenotyping and reviewed and approved the final manuscript; J.S. processed ThromboGenomics samples, performed deletion validations, and approved the final manuscript; I.G.T. contributed to data discussions and reviewed and approved the final manuscript; M.A.A. participated in the MDT, contributed to data discussions, and reviewed and approved the final manuscript; C.J.P. contributed to data discussions and reviewed and approved the final manuscript; W.H.O. contributed to data discussions and reviewed and approved the final manuscript; L.J. performed structural analysis, generated Figure 3A and associated text, contributed to data discussions, and reviewed and approved the final manuscript; and E.T. performed data analyses, developed the statistical models, contributed to data discussions, generated figures, coauthored the text, and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Claire L. Shovlin, Vascular Science, National Heart and Lung Institute, Imperial Centre for Translational and Experimental Medicine, Imperial College London, Hammersmith Campus, Du Cane Rd, London W12 0NN, United Kingdom; e-mail c.shovlin@imperial.ac.uk; and Ernest Turro, Department of Haematology, University of Cambridge, NHS Blood and Transplant, Long Rd, Cambridge CB4 3DF, United Kingdom; e-mail et341@cam.ac.uk.
