

Key Points
Vegfc deletion in endothelial or LepR+ cells compromises the bone marrow perivascular niche and hematopoietic stem cell maintenance.
Exogenously administered VEGF-C improves hematopoietic recovery after irradiation by accelerating endothelial and LepR+ cell regeneration.

Abstract
Hematopoietic stem cells (HSCs) reside in the bone marrow (BM) stem cell niche, which provides a vital source of HSC regulatory signals. Radiation and chemotherapy disrupt the HSC niche, including its sinusoidal vessels and perivascular cells, contributing to delayed hematopoietic recovery. Thus, identification of factors that can protect the HSC niche during an injury could offer a significant therapeutic opportunity to improve hematopoietic regeneration. In this study, we identified a critical function for vascular endothelial growth factor-C (VEGF-C), that of maintaining the integrity of the BM perivascular niche and improving BM niche recovery after irradiation-induced injury. Both global and conditional deletion of Vegfc in endothelial or leptin receptor–positive (LepR+) cells led to a disruption of the BM perivascular niche. Furthermore, deletion of Vegfc from the microenvironment delayed hematopoietic recovery after transplantation by decreasing endothelial proliferation and LepR+ cell regeneration. Exogenous administration of VEGF-C via an adenoassociated viral vector improved hematopoietic recovery after irradiation by accelerating endothelial and LepR+ cell regeneration and by increasing the expression of hematopoietic regenerative factors. Our results suggest that preservation of the integrity of the perivascular niche via VEGF-C signaling could be exploited therapeutically to enhance hematopoietic regeneration.
Introduction
BM niches for hematopoietic stem cells (HSCs) are specialized multicellular units that control HSC quiescence and self-renewal.1-3 The perivascular HSC niche is composed of endothelial cells (ECs) and leptin receptor–positive (LepR+) stromal cells, which produce factors that regulate hematopoietic stem and progenitor cells (HSPCs) in a paracrine manner.4-9
Radiation and chemotherapy disrupt the BM perivascular niche, leading to regression and remodeling of ECs and adipogenesis of LepR+ cells.10-15 HSC engraftment and proliferation after transplantation are supported by the BM microenvironment, including the perivascular niche.10,13,16-19 Thus, identification of factors that can protect the niche from irradiation damage or promote niche regeneration is of clinical interest and may improve HSC transplantation efficacy. VEGF-VEGFR2 signaling is crucial for HSC maintenance and endothelial regeneration after transplantation, but not for LepR+ cell maintenance.10,15,20 A previous report showed that VEGF-C, another VEGF family member and a major lymphangiogenic factor, regulates fetal erythropoiesis.21 VEGF-C is upregulated in BM ECs after sublethal irradiation.22 We thus hypothesized that VEGF-C could be an important factor in the perivascular niche, especially during BM regeneration.
We showed that VEGF-C maintains the LepR+ perivascular niche in the BM. Our results also revealed that loss of VEGF-C from the BM microenvironment delays vascular and hematopoietic recovery after transplantation. Viral vector–mediated delivery of VEGF-C promoted the perivascular niche and hematopoietic recovery from irradiation-induced damage, suggesting that VEGF-C has therapeutic potential.
Material and methods
Mice and tissues
Animal experiments were approved by the Committee for Animal Experiments of the District of Southern Finland. The mouse lines Vegfcflox/flox,23 Rosa26-CreERT2,24 Rosa26-LSL-tdTomato,25 Cdh5(BAC)-CreERT2,26 and Lepr-Cre27 have been described previously. All experiments were conducted on a C57BL/6 genetic background. B6.SJL mice were purchased from The Jackson Laboratory. C57BL/6J (RMS B.V.; Envigo, Blackthorn, UK) mice were used for studies employing adeno-associated viral vector (AAV). Cre-mediated recombination in adult mice was achieved by administering 5 consecutive intragastric doses of tamoxifen (20 mg/mL dissolved in 100 µL corn oil; Sigma-Aldrich, St. Louis, MO) to Vegfcflox/flox, Rosa26-CreERT2; Vegfcflox/flox (Cre+), and Cdh5(BAC)-CreERT2; Vegfcflox/flox (Cre+) mice. Adult C57bl/6J mice (8-10 weeks) received a single dose of a recombinant AAV serotype 9 (AAV9) encoding mVEGFR31-4-Ig28 (intraperitoneal [IP] injection of 1012 virus particles in 200 µL PBS), AAV9-derived FL-mVEGFC23 (IP; 1011 virus particles in 200 µL PBS), or VEGF-C protein.29 Control mice received AAVs that encoded domains 4 to 7 of VEGFR3-Ig, only the Fc domain, or mature VEGF-C with an inactivating (Asn163Arg) point mutation. Age- and sex-matched mice were used as controls.
Cell surface markers for hematopoietic stem and progenitor cells
The following cell surface markers were used: LKS (Lin−c-Kit+Sca1+), LT-HSC (FLT3−CD34−LKS or CD150+CD48−LKS), ST-HSC (FLT3−CD34+LKS), multipotential progenitor (MPP) (FLT3+CD34+LKS, or CD150−CD48−LKS), hematopoietic progenitor cell-1 (HPC-1) (CD150−CD48+LKS), and HPC-2 (CD150+CD48+LKS).
Flow cytometry and cell sorting
Whole bone marrow (WBM) cells were obtained by flushing tibias and femurs with HBSS (Hanks’ balanced salt solution; Ca2+- and Mg2+-free; Gibco; Thermo Fisher Scientific, Waltham, MA) supplemented with 2% fetal bovine serum. For niche cell analysis, mice were injected intravenously with 15 μg VE-cadherin-Alexa-647 (BioLegend, San Diego, CA) via tail vein 15 minutes before euthanasia. WBM was digested with collagenase I (Worthington Biochemical Corporation, Lakewood, NJ), Dispase II (Roche Applied Science, Basel, Switzerland), and DNase I (Sigma-Aldrich) in HBSS. Cells were resuspended in HBSS (Ca2+- and Mg2+-free) and 2% fetal bovine serum and stained using a 1:200 dilution of primary antibodies, unless otherwise indicated in the manufacturer's instructions. Analysis and sorting were performed on the FACSAria II flow cytometer. Data were analyzed with FlowJo v10 software (Tree Star Inc, Ashland OR).
scRNA-seq
For single-cell RNA sequencing (scRNA-seq), isolated CD45-Ter119-LepR-tdTomato–positive cells and CD45-Ter119-VE-cadherin–positive cells from Lepr-Cre;tdTomato BM were resuspended in 0.04% bovine serum albumin and HBSS and analyzed on the Chromium Single-Cell 3′RNA-sequencing system (10× Genomics, Pleasanton, CA) with Reagent Kit v2. Sample libraries were sequenced on the NovaSeq 6000 system equipped with the S1 flow cell (Illumina, San Diego, CA), with the following read lengths: 26 bp (read 1), 8 bp (i7 index), 0 bp (i5 index), and 91 bp (read 2).
CD45-Ter119-LepR+ cells and CD45-Ter119-VE-cadherin–positive cells purified from wild-type (WT), VciΔEC, and Vcfl/fl BM, using LepR and VE-cadherin antibodies, and lineage-CD45-Ter119- BM niche cells, isolated from irradiated/nonirradiated VciΔR26 and Vcfl/fl mice, were analyzed by using the Chromium Single-Cell 3′ Reagent Kit v3.1. Sample libraries were sequenced on the NovaSeq 6000 system with the S1 flow cell (Illumina), with the following read lengths: 28 bp (read 1), 8 bp (i7 index), 0 bp (i5 index), and 89 bp (read 2).
Real-time quantitative PCR
Total RNA was isolated by using the NucleoSpin RNA II Kit (Macherey-Nagel, Germany) or RNeasy Micro Kit (Qiagen, Hilden, Germany). Quantitative polymerase chain reaction (qPCR) was performed with either TaqMan Gene Expression Assays (Applied Biosystems, Thermo Fisher Scientific), accompanied by the iQTM Supermix kit, or SYBR Green oligos with the iQTM SYBR Green Supermix kit (Bio-Rad) on a C1000 Thermal cycler.
Mouse BM transplantation
Recipient mice were lethally irradiated 18 hours before transplantation with 10 Gy total-body irradiation (split doses, 2.5 hours apart) with the γ irradiator OB 29/4 (STS, Braunschweig, Germany).
Bone sectioning, immunofluorescence, and confocal microscopy
Freshly dissected femurs were fixed in 4% paraformaldehyde overnight at 4°C. Sections (100 μm thick) from the decalcified femurs were blocked in phosphate-buffered saline containing 0.3% Triton-X, 0.2% bovine serum albumin, 0.05% sodium azide, and 5% normal donkey serum for 1 hour and stained with primary antibodies for 2 days, followed by incubation with secondary antibodies. Immunofluorescence images were obtained with a LSM780 confocal microscope (20× numeric aperture [NA]:0.80; Zeiss, Oberkochen, Germany) or an Axioinvert microscope (5× NA:0.15;10× NA:0.3; Zeiss). Bright-field sections were viewed with a DM LB microscope (Leica Microsystems, Wetzlar, Germany), and images were captured with a DP50 color camera (Olympus Soft Imaging Solutions GmbH, Hamburg, Germany).
Statistical analysis
All bar graphs represent the mean ± SD. All data were derived from 2 to 4 individual experiments. For comparisons of 2 experimental groups, a 2-tailed Student t-test was used. For multiple comparisons, 1-way analysis of variance (Tukey’s multiple-comparisons test) was used (for all analyses, Prism 8.0; GraphPad was used).
More information is provided in “Detailed Experimental Procedures” in the supplemental Data (available on the Blood Web site).
Results
VEGF-C is expressed in LepR+ cells and EC subsets in the BM
VEGF-C expression has been reported in hematopoietic cells and ECs in the developing embryo21,30 and in adult smooth muscle31 and osteolineage cells.32 To define the cell types in the adult mouse BM that produce VEGF-C, we first analyzed Vegfc mRNA level in BM cells by using qPCR. We found that Vegfc expression was higher in nonhematopoietic LepR+ cells and PECAM-1+ cells than in WBM (Figure 1A). We then performed scRNA-seq on Lepr-tdTomato–positive cells and VE-cadherin–positive ECs isolated from the BM of Lepr-Cre;tdTomato mice (Figure 1B). Graph-based clustering was performed for ∼6000 single-cell transcriptomes and visualized with Uniform Manifold Approximation and Projection (UMAP; Figure 1C). These clusters were (1) 2 arterial EC (AEC) subsets (Ly6a arteriolar EC cluster and Ly6a/Cd34 AEC cluster); (2) 2 sinusoidal EC clusters (Stab-2 SEC-1 and Stab2/Klf4hi SEC-2); (3) 5 LepR subsets (Lplhi Lepr-1 cluster; Wisp2hi Lepr-2 cluster; Cxcl14hi Lepr-3 cluster; osteolineage-primed Lepr-4 cells expressing Wif1 and Bgalp2; and fibroblast lineage Lepr-5 cells expressing Aspn and Col3a1 (Figure 1C; supplemental Figure 1A). Cell projection using singleR33 and 3 different published BM niche scRNA data sets34-36 as references confirmed these cell identities in our Seurat37,38 analysis (supplemental Figure 1B-G). LepR-expressing cells, AECs, and some chondrocytes, SECs, and fibroblasts expressed VEGF-C, as shown in our data set and the 3 published reference data sets34-36 (Figure 1D; supplemental Figure 1B-D). Two of the main receptors for VEGF-C, VEGFR-3 (Flt4), and VEGFR-2 (Kdr) were expressed in the ECs (Figure 1D). Thus, both VEGF-C and its receptors are expressed in the adult BM perivascular niche.

VEGF-C regulates the integrity of the BM LepR+ HSC niche. (A) Relative Vegfc mRNA level in sorted WT PECAM-1+ ECs and LepR+ stromal cells, analyzed by qPCR (normalized to WBM; n = 2 mice per group). (B) Experimental outline of scRNA-seq analysis of LepR+ cells and VE-cadherin–positive ECs from Lepr-Cre;tdTomato BM. (C) UMAP plots of BM LepR-tdTomato–positive cells and VE-cadherin–positive ECs (left). A feature plot shows Lepr and Cdh5 expression (right). (D) A feature plot showing Vegfc, Flt4 (VEGFR-3), and Kdr (VEGFR-2) expression. (E) Experimental setup for evaluating the effects of Vegfc deletion in the BM. Vegfc was deleted from adult BM by administering 5 daily tamoxifen injections to 7- to 10-week-old Rosa26-CreERT2;Vegfcflox/flox mice. (F) Representative confocal immunofluorescence images of femur sections from VciΔR26 and Vcfl/fl mice 5 months after deletion. Staining for ECs (endomucin, green) and perivascular cells (LepR, red), with quantification (right) (n = 5-6 mice per group). Bar represents 50 μm. (G) Quantification of HSPC subsets per femur from 7-month-old VciΔR26 mice and Vcfl/fl littermate controls, determined by CD48 and CD150 staining (n = 13-14 mice per group). Quantification of HSPC subsets per femur from 22- to 23-month-old VciΔR26 mice and Vcfl/fl littermate controls (n = 6 male mice per group). Values show mean ± SD. Statistical significance was determined with the 2-tailed, unpaired Student t test. MPP, multipotential progenitor. *P < .05; **P < .01.
VEGF-C regulates the integrity of the BM LepR+ HSC niche. (A) Relative Vegfc mRNA level in sorted WT PECAM-1+ ECs and LepR+ stromal cells, analyzed by qPCR (normalized to WBM; n = 2 mice per group). (B) Experimental outline of scRNA-seq analysis of LepR+ cells and VE-cadherin–positive ECs from Lepr-Cre;tdTomato BM. (C) UMAP plots of BM LepR-tdTomato–positive cells and VE-cadherin–positive ECs (left). A feature plot shows Lepr and Cdh5 expression (right). (D) A feature plot showing Vegfc, Flt4 (VEGFR-3), and Kdr (VEGFR-2) expression. (E) Experimental setup for evaluating the effects of Vegfc deletion in the BM. Vegfc was deleted from adult BM by administering 5 daily tamoxifen injections to 7- to 10-week-old Rosa26-CreERT2;Vegfcflox/flox mice. (F) Representative confocal immunofluorescence images of femur sections from VciΔR26 and Vcfl/fl mice 5 months after deletion. Staining for ECs (endomucin, green) and perivascular cells (LepR, red), with quantification (right) (n = 5-6 mice per group). Bar represents 50 μm. (G) Quantification of HSPC subsets per femur from 7-month-old VciΔR26 mice and Vcfl/fl littermate controls, determined by CD48 and CD150 staining (n = 13-14 mice per group). Quantification of HSPC subsets per femur from 22- to 23-month-old VciΔR26 mice and Vcfl/fl littermate controls (n = 6 male mice per group). Values show mean ± SD. Statistical significance was determined with the 2-tailed, unpaired Student t test. MPP, multipotential progenitor. *P < .05; **P < .01.
VEGF-C regulates the integrity of the BM LepR+ perivascular niche
To understand the functional significance of VEGF-C signaling on the BM perivascular niche and hematopoiesis, we deleted Vegfc from 7- to 10-week-old Vegfc conditional knockout mice, using tamoxifen-inducible Rosa26-CreERT2 recombinase (VciΔR26; Figure 1E; supplemental Figure 2A). By 7 months of age, immunofluorescence revealed a significantly smaller LepR+ BM area in VciΔR26 mice than in Vcfl/fl mice, whereas the endomucin-positive vessel area was not significantly altered (Figure 1F). Flow cytometry confirmed the decrease in LepR+ cells in relation to the PECAM-1–stained ECs, which were not altered (supplemental Figure 2B-C). A decrease in BM LepR+ cells in the absence of VEGF-C was recapitulated in WT mice in which VEGF-C was inactivated by the “VEGF-C trap,” involving AAV delivery of soluble mVEGFR-31-4-Ig28 (supplemental Figure 2J). These data suggest that VEGF-C is important for regulating the composition of the perivascular niche by maintaining the LepR+ cells.
Assessment of hematopoietic parameters revealed that the VciΔR26 mice at 7 months of age had normal BM cellularity, spleen size, white blood cell (WBC) counts, BM lineage composition, and HSPC subtypes when analyzed with SLAM surface markers (CD48 and CD150; Figure 1G; supplemental Figure 2D-F). Similar results were obtained in an analysis of the number of HSPCs, with CD34 and FLT3 used as alternative surface markers (supplemental Figure 2G-I). However, by 22 to 23 months of age, the VciΔR26 mice had developed mild leukopenia and a significant reduction in HSCs and methylcellulose colony-forming cells, despite their relatively intact BM cellularity and lineage composition (Figure 1G; supplemental Figure 2K-O). These data suggest that a prolonged disruption of the perivascular niche because of VEGF-C loss compromises the long-term maintenance of normal hematopoiesis.
VEGF-C from a subset of ECs contributes to BM LepR+ cell maintenance
Because the scRNA analysis indicated that both BM LepR+ cells and ECs are sources of VEGF-C in the adult BM microenvironment, we crossed Vegfcfl/fl mice with the corresponding cell-type–specific deleter mice. To evaluate whether ECs provide a functionally significant source of VEGF-C to support the BM perivascular niche, we conditionally deleted Vegfc from adult BM ECs in Cdh5(BAC)-CreERT2;Vegfcfl/fl (VciΔEC) mice. Immunostaining for LepR and endomucin in femur sections and flow cytometry of LepR+ cells documented a significant decrease in LepR+ cells in VciΔEC mice as well, whereas the ECs were not significantly altered (Figure 2A-B; supplemental Figure 3A-B). Deletion of endothelial Vegfc resulted in a small but significant decrease in BM HSCs, whereas BM cellularity, spleen size, WBC counts, and BM lineage composition were not significantly altered (Figure 2C; supplemental Figure 3C-F).
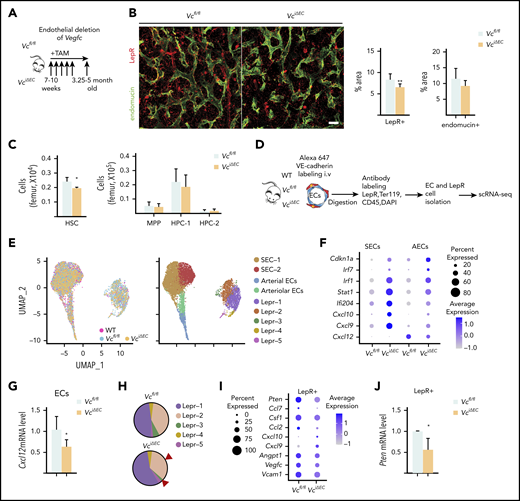
Endothelial cells serve as a functionally significant source of VEGF-C in the BM. (A) Experimental setup for evaluating the effects of Vegfc deletion in ECs. Vegfc was deleted from ECs in 7- to 10-week-old Cdh5-CreERT2;Vegfcflox/flox mice by tamoxifen injections. (B) Representative confocal immunofluorescence images of femur sections from VciΔEC mice and their littermate controls stained for ECs (endomucin, green) and LepR+ perivascular cells (red), with quantifications (right) (n = 6 mice per group). Bar represents 50 μm. (C) Quantification of HSPC subsets per femur from Vcfl/fl and VciΔEC mice determined by CD48 and CD150 staining (n = 13-14). (D) Experimental outline of scRNA-seq analysis of LepR+ cells and VE-cadherin–positive ECs from WT, Vcfl/fl, and VciΔEC mice. (E) UMAP plot of integrated BM ECs and LepR+ cells isolated from WT, Vcfl/fl, and VciΔEC mice (left). UMAP plots showing the clustering of integrated BM ECs and LepR+ cells from WT, Vcfl/fl, and VciΔEC mice (right). (F) Dot plot showing selected differentially expression genes in SECs (SEC-1 and -2), AECs, and arteriolar ECs after endothelial Vegfc deletion in comparison with Vcfl/fl mice. (G) Relative Cxcl12 mRNA levels in isolated BM ECs from VciΔEC and littermate control mice analyzed by qPCR. (H) Comparison of relative cell counts in Lepr-1, -2, -3, -4, and -5 clusters from Vcfl/fl and VciΔEC mice. (I) Dot plot showing selected differentially expression genes in LepR+ cells (Lepr-1, -2, and -3) after endothelial Vegfc deletion in comparison with the Vcfl/fl mice. (J) Relative Pten mRNA levels in isolated BM LepR+ cells from VciΔEC and littermate control mice analyzed by qPCR. *P < .05; **P < .01. MPP, multipotential progenitor.
Endothelial cells serve as a functionally significant source of VEGF-C in the BM. (A) Experimental setup for evaluating the effects of Vegfc deletion in ECs. Vegfc was deleted from ECs in 7- to 10-week-old Cdh5-CreERT2;Vegfcflox/flox mice by tamoxifen injections. (B) Representative confocal immunofluorescence images of femur sections from VciΔEC mice and their littermate controls stained for ECs (endomucin, green) and LepR+ perivascular cells (red), with quantifications (right) (n = 6 mice per group). Bar represents 50 μm. (C) Quantification of HSPC subsets per femur from Vcfl/fl and VciΔEC mice determined by CD48 and CD150 staining (n = 13-14). (D) Experimental outline of scRNA-seq analysis of LepR+ cells and VE-cadherin–positive ECs from WT, Vcfl/fl, and VciΔEC mice. (E) UMAP plot of integrated BM ECs and LepR+ cells isolated from WT, Vcfl/fl, and VciΔEC mice (left). UMAP plots showing the clustering of integrated BM ECs and LepR+ cells from WT, Vcfl/fl, and VciΔEC mice (right). (F) Dot plot showing selected differentially expression genes in SECs (SEC-1 and -2), AECs, and arteriolar ECs after endothelial Vegfc deletion in comparison with Vcfl/fl mice. (G) Relative Cxcl12 mRNA levels in isolated BM ECs from VciΔEC and littermate control mice analyzed by qPCR. (H) Comparison of relative cell counts in Lepr-1, -2, -3, -4, and -5 clusters from Vcfl/fl and VciΔEC mice. (I) Dot plot showing selected differentially expression genes in LepR+ cells (Lepr-1, -2, and -3) after endothelial Vegfc deletion in comparison with the Vcfl/fl mice. (J) Relative Pten mRNA levels in isolated BM LepR+ cells from VciΔEC and littermate control mice analyzed by qPCR. *P < .05; **P < .01. MPP, multipotential progenitor.
VEGFR-2 and -3 were expressed in BM ECs but not in LepR+ cells (Figure 1D), suggesting that VEGF-C from ECs may indirectly regulate LepR+ cells in the perivascular niche. Although we did not observe significant phenotypic changes in the ECs, we suspected that signaling in ECs was altered after Vegfc deletion and may have been responsible for the loss of LepR+ cells in the VciΔEC mice. Thus, we performed scRNA analysis of BM ECs and LepR+ cells isolated from Vcfl/fl and VciΔEC mice by flow cytometry (Figure 2D-E; supplemental Figure 4A,C). There were no significant changes in the percentages of BM SEC-1 and -2, arteriolar or arterial ECs in the mice when analyzed in Seurat-annotated EC subclusters (supplemental Figure 4B). Nevertheless, analysis of differentially expressed genes in endothelial clusters between VciΔEC mice and Vcfl/fl mice indicated that the expression of cell cycle inhibitor (Cdkn1a) and inflammation-related genes was upregulated in ECs from VciΔEC mice, whereas the hematopoietic niche factor Cxcl125-8 was downregulated (Figure 2F; supplemental Figure 4D). We confirmed the decrease in Cxcl12 in the Vegfc-deleted ECs by qPCR analysis (Figure 2G). Functional annotation clustering analysis using the Database for Annotation, Visualization, and Integrated Discovery (DAVID)39,40 and gene set enrichment analysis revealed that the biological processes associated with the genes upregulated in endothelial subclusters from VciΔEC mice in comparison with Vcfl/fl mice were related to cellular response to interferon signaling and immune system response, whereas the downregulated genes were associated with triglyceride and cholesterol homeostasis (supplemental Figure 4E-G). This finding is consistent with a report showing that VEGF-C–mediated stimulation of VEGFR-3 in lymphatic ECs leads to the downregulation of genes involved in interferon responses.41
In addition to reduced LepR+ cellularity, scRNA-seq showed specific reduction in cells in Lepr-2 and -3 clusters in VciΔEC mice; these represent LepR+ cells closely related to SECs34 (Figure 2H). Differential gene expression analysis of the LepR+ cells in the scRNA-seq data indicated that Pten was decreased in LepR+ cells, which was confirmed by qPCR analysis of isolated LepR+ cells (Figure 2I-J; supplemental Figure 4H-I). Pten has been shown to regulate the maintenance of LepR+ cells.42 The decreased Pten level in LepR+ cells may thus contribute to the decrease in LepR+ cells in the BM. Niche factors Vcam1, Angpt1, and Csf1 were also decreased in LepR+ cells from VciΔEC mice, whereas some inflammation-related chemokine genes, for example Cxcl9 and Cxcl10, were upregulated (Figure 2I; supplemental Figure 4H).
These data indicate that loss of VEGF-C from the ECs significantly impairs the expression of critical HSC-supporting genes in ECs and LepR+ cells. VEGF-C produced by ECs thus contributes to the normal endothelial program that supports LepR+ cells and HSCs.
VEGF-C from LepR+ cells contributes to BM vascular development and perivascular niche maintenance
To investigate whether LepR+ cell–derived VEGF-C also contributes to perivascular niche integrity, we deleted Vegfc from LepR+ cells using the Lepr-Cre (VcΔLepr) allele, which becomes active right after birth.42 A decrease in BM LepR+ areas and in diaphyseal endomucin-positive ECs was observed in femur sections of 7-week-old VcΔLepr mice. This result was consistent with reduction in the VE-cadherin– and LepR-expressing cells observed in flow cytometry analysis (Figure 3A-B; supplemental Figure 5A-B). The proliferation of ECs was decreased already in 4-week-old VcΔLepr mice, which may explain the stronger vascular defect in these mice (supplemental Figure 5C) than in mice in which Vegfc was deleted in adulthood (Figures 1 and 2). This finding suggests that the early loss of VEGF-C from the LepR+ cells compromises, not only LepR+ cells in the perivascular niche, but also vascular development.
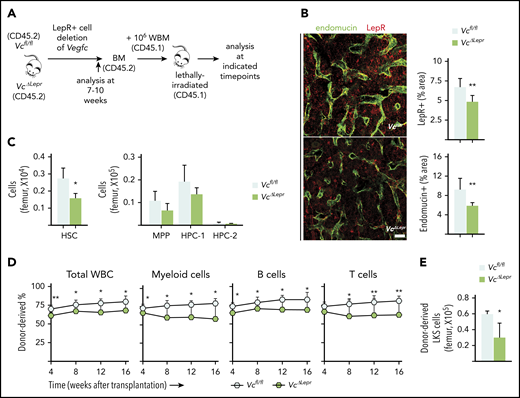
LepR+ cell–derived VEGF-C contributes to maintenance of functional HSCs in the BM. (A) Experimental setup for evaluating the effects of LepR+ cell–derived VEGF-C. Vegfc was deleted from LepR+ cells using Lepr-Cre. BM of 7- to 10-week-old mice was analyzed for niche cell and HSC phenotypes. WBM from 10-week-old VcΔLepr or Vcfl/fl mice was transplanted competitively with CD45.1 WBM into lethally irradiated CD45.1 mice. (B) Representative confocal immunofluorescence images of femur sections from VcΔLepr and Vcfl/fl littermate controls stained for ECs (endomucin, green) and LepR+ perivascular cells (red), with quantification (right) (n = 6-7 mice per group). Bar represents 50 μm. (C) Quantification of HSPC subsets per femur from VcΔLepr and Vcfl/fl littermate control mice (n = 4 mice per group) determined by CD48 and CD150 staining. (D) Competitive transplantation of WBM from VcΔLepr and their Vcfl/fl littermate control mice into lethally irradiated WT CD45.1 recipients (2 independent transplants with 3 to 4 recipients per condition per transplant). Shown is a multilineage donor chimerism from peripheral blood at the indicated time points after competitive transplantation. (E) Quantification of LKS cells derived from VcΔLepr or Vcfl/fl mouse BM 16 weeks after transplantation. Values show means ± SD. Statistical significance was determined using the 2-tailed, unpaired Student t test. *P < .05; **P < .01.
LepR+ cell–derived VEGF-C contributes to maintenance of functional HSCs in the BM. (A) Experimental setup for evaluating the effects of LepR+ cell–derived VEGF-C. Vegfc was deleted from LepR+ cells using Lepr-Cre. BM of 7- to 10-week-old mice was analyzed for niche cell and HSC phenotypes. WBM from 10-week-old VcΔLepr or Vcfl/fl mice was transplanted competitively with CD45.1 WBM into lethally irradiated CD45.1 mice. (B) Representative confocal immunofluorescence images of femur sections from VcΔLepr and Vcfl/fl littermate controls stained for ECs (endomucin, green) and LepR+ perivascular cells (red), with quantification (right) (n = 6-7 mice per group). Bar represents 50 μm. (C) Quantification of HSPC subsets per femur from VcΔLepr and Vcfl/fl littermate control mice (n = 4 mice per group) determined by CD48 and CD150 staining. (D) Competitive transplantation of WBM from VcΔLepr and their Vcfl/fl littermate control mice into lethally irradiated WT CD45.1 recipients (2 independent transplants with 3 to 4 recipients per condition per transplant). Shown is a multilineage donor chimerism from peripheral blood at the indicated time points after competitive transplantation. (E) Quantification of LKS cells derived from VcΔLepr or Vcfl/fl mouse BM 16 weeks after transplantation. Values show means ± SD. Statistical significance was determined using the 2-tailed, unpaired Student t test. *P < .05; **P < .01.
The VcΔLepr mice showed a decreased number of HSCs and reduced methylcellulose colony-forming ability at 7 weeks of age (Figure 3C; supplemental Figure 5E), whereas the BM cellularity, spleen size, WBC counts, and BM lineage composition were not significantly altered at this age (supplemental Figure 5D-G). When VcΔLepr BM was transplanted competitively into lethally irradiated WT recipients, significantly impaired long-term multilineage reconstitution was observed in all peripheral blood lineages between 8 and 16 weeks after transplantation (Figure 3D). The recipients of VcΔLepr BM also showed a reduction in donor-derived LKS cells in the BM at 16 weeks after transplantation (Figure 3E). Furthermore, we observed a reduced colony forming ability in CD150+CD48− HSCs from VcΔLepr mice in primary methylcellulose cultures (supplemental Figure 5H), indicating their compromised functional properties. As with endothelial-specific Vegfc deletion, a decrease in Pten level was also observed in LepR+ cells from the VcΔLepr mice (supplemental Figure 5I). These data suggest that VEGF-C produced by LepR+ cells is essential for BM vascular development, maintenance of LepR+ cells in the perivascular niche, and maintenance of a functional HSC pool.
Loss of VEGF-C in BM stroma delays vascular and HSC regeneration after irradiation
Consistent with published data,22 we found that irradiation causes an increase in Vegfc mRNA expression in the BM. Twenty-four hours after lethal irradiation, a ∼100-fold increase in Vegfc mRNA was observed in BM nonhematopoietic cells, suggesting that the microenvironment acts as the predominant source of VEGF-C in the postirradiation marrow (Figure 4A). VEGF-C protein was also increased in WBM 1 day after irradiation (supplemental Figure 6A). Thus, we reasoned that the increase in VEGF-C after irradiation may be important for perivascular niche and hematopoietic recovery.
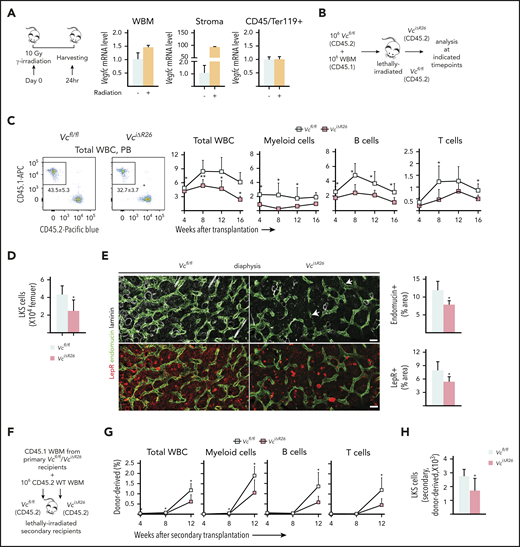
Loss of VEGF-C from the BM microenvironment delays vascular and HSC regeneration after irradiation. (A) Experimental setup for evaluating Vegfc expression level in the BM after irradiation, by using qPCR. Relative Vegfc mRNA level in WBM, in sorted CD45−Ter119− stromal cells, and in CD45+Ter119+ hematopoietic cells 24 hours after 10-Gy radiation (normalized to untreated WBM; n = 2 mice). (B) Experimental setup for evaluating the efficiency of engraftment of WT WBM in lethally irradiated VciΔR26 and Vcfl/fl mice. (C) Transplantation of WT WBM (CD45.1) into lethally irradiated VciΔR26 mice and their Vcfl/fl littermate hosts. Representative flow cytometry graph of total WBCs from peripheral blood 12 weeks after transplantation (left). The kinetics of multilineage donor chimerism from peripheral blood after transplantation (right; n = 6-8 mice per group). (D) Quantification of BM LKS cells in lethally irradiated VciΔR26 mice and their Vcfl/fl littermate recipients 16 weeks after transplantation (n = 6-8 mice per group). (E) Representative confocal immunofluorescence images and quantification. Femur sections from VciΔR26 mice and their littermate controls stained for ECs and basement membranes (endomucin, green; laminin, white) and LepR cells (red) (n = 4-5 mice per group). Bars represent 50 μm. (F) Experimental setup for serial transplantation. WBM (CD45.1) from lethally irradiated primary VciΔR26 and Vcfl/fl recipients was transplanted competitively with CD45.2 WBM into lethally irradiated secondary VciΔR26 and Vcfl/fl recipients. (G) Multilineage donor chimerism from peripheral blood after secondary competitive transplantation (2 independent transplants with 3 recipients per condition per transplant). (H) Quantification of BM LKS numbers in secondary VciΔR26 mice and their Vcfl/fl littermate controls 16 weeks after transplantation. Reported values are mean ± SD. Statistical significance was determined using the 2-tailed, unpaired Student t test. *P < .05; **P < .01.
Loss of VEGF-C from the BM microenvironment delays vascular and HSC regeneration after irradiation. (A) Experimental setup for evaluating Vegfc expression level in the BM after irradiation, by using qPCR. Relative Vegfc mRNA level in WBM, in sorted CD45−Ter119− stromal cells, and in CD45+Ter119+ hematopoietic cells 24 hours after 10-Gy radiation (normalized to untreated WBM; n = 2 mice). (B) Experimental setup for evaluating the efficiency of engraftment of WT WBM in lethally irradiated VciΔR26 and Vcfl/fl mice. (C) Transplantation of WT WBM (CD45.1) into lethally irradiated VciΔR26 mice and their Vcfl/fl littermate hosts. Representative flow cytometry graph of total WBCs from peripheral blood 12 weeks after transplantation (left). The kinetics of multilineage donor chimerism from peripheral blood after transplantation (right; n = 6-8 mice per group). (D) Quantification of BM LKS cells in lethally irradiated VciΔR26 mice and their Vcfl/fl littermate recipients 16 weeks after transplantation (n = 6-8 mice per group). (E) Representative confocal immunofluorescence images and quantification. Femur sections from VciΔR26 mice and their littermate controls stained for ECs and basement membranes (endomucin, green; laminin, white) and LepR cells (red) (n = 4-5 mice per group). Bars represent 50 μm. (F) Experimental setup for serial transplantation. WBM (CD45.1) from lethally irradiated primary VciΔR26 and Vcfl/fl recipients was transplanted competitively with CD45.2 WBM into lethally irradiated secondary VciΔR26 and Vcfl/fl recipients. (G) Multilineage donor chimerism from peripheral blood after secondary competitive transplantation (2 independent transplants with 3 recipients per condition per transplant). (H) Quantification of BM LKS numbers in secondary VciΔR26 mice and their Vcfl/fl littermate controls 16 weeks after transplantation. Reported values are mean ± SD. Statistical significance was determined using the 2-tailed, unpaired Student t test. *P < .05; **P < .01.
To evaluate the effect of microenvironment-derived VEGF-C on the hematopoietic niche and HSC regeneration upon injury, we first performed transplantation of WT cells into lethally irradiated VciΔR26 recipient mice. Impaired long-term multilineage reconstitution was observed in VciΔR26 recipients, as evidenced by significantly decreased donor-derived reconstitution in all peripheral WBC lineages between 8 and 16 weeks after transplantation. Donor-derived LKS cells were also reduced in the BM 16 weeks after transplantation (Figure 4B-D). Decreases in endomucin-positive and LepR+ areas, as well as declining endothelial proliferation, was observed in the BM of irradiated VciΔR26-recipient mice 16 weeks after transplantation (Figure 4E; supplemental Figure 6B), indicating that VEGF-C promoted EC proliferation during vascular regeneration. Moreover, some vessels in VciΔR26 recipients lacked basement-membrane laminin coverage (Figure 4E). These data indicate that lack of VEGF-C during irradiation-induced injury compromises the recovery of BM vascular and perivascular components and hematopoietic regeneration.
To evaluate the possibility that the defects attributed to hematopoietic recovery upon loss of niche-derived VEGF-C are caused by impaired homing of HSCs, we labeled WT WBM cells with CFSE and transplanted them into lethally irradiated VciΔR26 and Vcfl/fl mice. We did not find a significant difference in the number of CFSE-labeled LKS cells between VciΔR26 and Vcfl/fl recipients 16 hours after transplantation (supplemental Figure 6C).
To evaluate the effect of niche-derived VEGF-C on HSC self-renewal in the transplantation setting, we used serial transplantation experiments, with VciΔR26 and Vcfl/fl mice as both primary and secondary recipients (Figure 4F). These experiments demonstrated an impaired multilineage reconstitution in VciΔR26 recipients and a reduced number of donor-derived LKS cells 12 weeks after secondary transplantation (Figure 4G-H). These data suggest that microenvironment-derived VEGF-C is necessary for HSC self-renewal after transplantation.
To investigate the cellular source of the VEGF-C that is necessary for niche regeneration after injury, we performed transplantation experiments in VcΔLepr and VciΔEC mice. An impaired long-term multilineage reconstitution of WT BM cells, reduced the number of LKS cells, and decreases in both LepR+ and endomucin-positive areas were observed in irradiated VcΔLepr and VciΔEC recipients (supplemental Figure 6D-H). In summary, our data suggest that VEGF-C from both ECs and LepR+ cells contributes to niche regeneration after irradiation, which in turn affects hematopoietic recovery and HSC self-renewal after transplantation.
Exogenous VEGF-C improves BM recovery after irradiation-induced damage
In light of our findings that niche-derived VEGF-C contributes to niche regeneration and hematopoietic recovery upon transplantation, we investigated whether providing exogenous VEGF-C would have a therapeutic advantage in the recovery from radiation-induced damage. To determine whether pretreatment with VEGF-C can mitigate the effects of irradiation-induced injury in the BM, we injected AAV9 encoding mouse VEGF-C or control protein systemically into WT mice and sublethally irradiated the mice 7 days later with a single 4-Gy dose (Figure 5A). We confirmed that AAV9 induced an increase in Vegfc mRNA in the liver (supplemental Figure 7A). Seven days after radiation, we observed significantly more ECs and LepR+ cells in the irradiated mice that were pretreated with AAV9-derived VEGF-C than in mice that expressed Fc control or inactive VEGF-C (Figure 5B; supplemental Figure 7B). Moreover, the mice showed an increase in BM cellularity and LKS cells (Figure 5C-D; supplemental Figure 7C).

VEGF-C improves BM recovery after irradiation-induced damage. (A) Experimental setup for evaluating the effects of exogenous VEGF-C upon irradiation-induced injury. AAV9 encoding mouse VEGF-C or control protein was injected systemically (IP) to WT mice 7 days before irradiation. BM was analyzed 7 days after irradiation. (B) Quantification of BM VE-cadherin–positive ECs and LepR+ cells in VEGF-C–treated WT mice 7 days after 4-Gy irradiation by flow cytometry (n = 6-8 individual mice per group). (C) Quantification of LKS cells per femur (n = 6-8 mice per group) by flow cytometry. (D) Hematoxylin and eosin staining of femur sections 7 days after 4-Gy irradiation (n = 6-8 mice per group). Bar represents 50 μm. (E) Experimental setup for evaluating the effects of exogenous VEGF-C on the efficiency of hematopoietic engraftment after transplantation. AAV9 encoding mouse VEGF-C or control protein was injected systemically into WT mice 1 day after lethal irradiation and BM transplantation. BM was analyzed 2 and 3 weeks after transplantation. (F-G) Flow cytometry quantification of BM ECs, LepR+ cells, and LKS cells in mice treated with VEGF-C after lethal irradiation and BM transplantation (n = 3 per group per time point). Values show the mean ± SD. Statistical significance was determined using the Student t test or 1-way analysis of variance multiple-comparisons test. *P < .05; **P < .01; ***P < .001.
VEGF-C improves BM recovery after irradiation-induced damage. (A) Experimental setup for evaluating the effects of exogenous VEGF-C upon irradiation-induced injury. AAV9 encoding mouse VEGF-C or control protein was injected systemically (IP) to WT mice 7 days before irradiation. BM was analyzed 7 days after irradiation. (B) Quantification of BM VE-cadherin–positive ECs and LepR+ cells in VEGF-C–treated WT mice 7 days after 4-Gy irradiation by flow cytometry (n = 6-8 individual mice per group). (C) Quantification of LKS cells per femur (n = 6-8 mice per group) by flow cytometry. (D) Hematoxylin and eosin staining of femur sections 7 days after 4-Gy irradiation (n = 6-8 mice per group). Bar represents 50 μm. (E) Experimental setup for evaluating the effects of exogenous VEGF-C on the efficiency of hematopoietic engraftment after transplantation. AAV9 encoding mouse VEGF-C or control protein was injected systemically into WT mice 1 day after lethal irradiation and BM transplantation. BM was analyzed 2 and 3 weeks after transplantation. (F-G) Flow cytometry quantification of BM ECs, LepR+ cells, and LKS cells in mice treated with VEGF-C after lethal irradiation and BM transplantation (n = 3 per group per time point). Values show the mean ± SD. Statistical significance was determined using the Student t test or 1-way analysis of variance multiple-comparisons test. *P < .05; **P < .01; ***P < .001.
To test whether administering VEGF-C protein after irradiation has a similar positive effect, we first irradiated mice sublethally with 4 Gy and, 24 hours later, started daily IP injections of 1.25 mg/kg VEGF-C for 3 days. We found that the 3-day treatment with VEGF-C protein was sufficient to increase the percentage of LKS+ cells in the BM (supplemental Figure 7D). These data suggest that administering VEGF-C after radiation damage can also improve BM recovery.
Next, we investigated whether administration of VEGF-C would improve hematopoietic recovery upon BM transplantation after lethal irradiation. We systemically injected AAV9 encoding mouse VEGF-C or control protein into WT mice 1 day after lethal irradiation by using 10 Gy plus BM transplantation (Figure 5E; supplemental Figure 7E). We did not observe significant changes in BM cellularity, peripheral WBC counts, or spleen size at either 2 or 3 weeks after transplantation (supplemental Figure 7F-G). A transient decrease in the BM B-cell percentage and a corresponding transient increase in the myeloid cell percentage was observed at 2 weeks after transplantation in mice that were overexpressing VEGF-C (supplemental Figure 7H). A significant increase in ECs and donor-derived LKS cells was observed at both 2 and 3 weeks after transplantation, whereas LepR+ cells were significantly increased by 3 weeks (Figure 5F-G). These results suggest that administering VEGF-C after irradiation and transplantation can improve the vascular and perivascular niche recovery as well as hematopoietic regeneration.
VEGF-C promotes EC proliferation and the expression of hematopoietic-regenerative factors after irradiation
To understand the mechanism of VEGF-C mitigation of radiation-induced damage in the BM niche cells, we isolated the BM nonhematopoietic cell fraction from 4-Gy–irradiated and –nonirradiated Vcfl/fl and VciΔR26 mice for scRNA-seq analysis 7 days after irradiation. Graph-based clustering was performed on ∼6000 single-cell transcriptomes of the nonhematopoietic niche cells from the BM, and the results were visualized with UMAP (Figure 6A-B; supplemental Figure 8A-D). We observed 3 new EC clusters associated with irradiation (damaged ECs, Aqp1hi ECs, Cxcl10 ECs; Figure 6B-C). The damaged ECs were characterized by the lack of specific marker transcripts and upregulation of ribosomal gene expression. Aqp1hi ECs were characterized by lower expression of SEC-2 markers and higher expression in Aqp1, Ackr1, and Plvap (supplemental Figure 8C). Damaged ECs and Aqp1hi ECs emerged after irradiation, predominantly in irradiated VciΔR26 mice (Figure 6D). Cxcl10 ECs were decreased in nonirradiated VciΔR26 mice vs Vcfl/fl mice, but increased in irradiated VciΔR26 mice. After irradiation, there were more ECs in the S phase, whereas Vegfc deletion decreased the proportion of proliferating ECs. In agreement with this, we observed a reduction in Pcna expression in the irradiated VciΔR26 ECs (Figure 6E). Notch receptors (Notch1 and Notch4), ligand (Dll4), and a target gene (Hes1), which are involved in hematopoietic maintenance and regeneration34,43,44 were decreased in irradiated VciΔR26 EC (Figure 6E). Many hematopoietic regenerative genes, such as Angpt2/Tek, Sema3f, and Thbd,6,13,22,44,45 and the VEGF-C receptors Flt4 and Kdr were upregulated in irradiated ECs, but not in VciΔR26 ECs in response to irradiation (Figure 6E). In addition, the irradiation further increased the expression of the inflammatory molecules S100a8 and S100a9 in VciΔR26 ECs (Figure 6E).
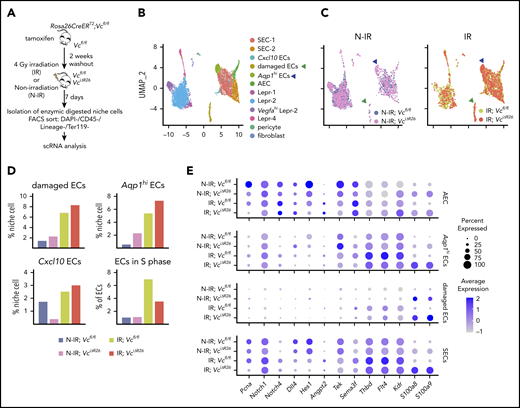
VEGF-C promotes BM EC proliferation and increases the expression of hematopoietic-regenerative factors after irradiation. (A) Experimental setup for evaluating the effects of Vegfc in the BM upon irradiation. Vegfc was deleted from the BM in 10-week-old Rosa26-CreERT2;Vegfcflox/flox mice using tamoxifen injections. VciΔR26 and Vcfl/fl mice received 4-Gy irradiation after a 2-week tamoxifen washout. Niche cells were isolated from irradiated (IR) VciΔR26 and Vcfl/fl mice and their nonirradiated (N-IR) controls 7 days after irradiation and analyzed using scRNA-seq. (B-C) UMAP plot showing the clustering of integrated nonhematopoietic BM stroma cells from IR VciΔR26 and Vcfl/fl mice and their N-IR controls. UMAP plot showing nonhematopoietic BM stroma cells from irradiated VciΔR26 and Vcfl/fl mice and their N-IR controls separately. Note that damaged ECs (green arrowhead) and Aqp1hi ECs (blue arrowhead) are increased after irradiation. (D) Percentages of the clusters (damaged ECs, Aqp1hi ECs, and Cxcl10 ECs) and quantification of ECs in S phase. (E) Dot plot showing selected differentially expressed genes in SECs (SEC-1, SEC-2, and Cxcl10 ECs), damaged ECs, Aqp1hi ECs, and AECs. Analysis of differentially expressed genes was performed between IR;Vcfl/fl vs N-IR;Vcfl/fl and IR;VciΔR26 vs N-IR;VciΔR26.
VEGF-C promotes BM EC proliferation and increases the expression of hematopoietic-regenerative factors after irradiation. (A) Experimental setup for evaluating the effects of Vegfc in the BM upon irradiation. Vegfc was deleted from the BM in 10-week-old Rosa26-CreERT2;Vegfcflox/flox mice using tamoxifen injections. VciΔR26 and Vcfl/fl mice received 4-Gy irradiation after a 2-week tamoxifen washout. Niche cells were isolated from irradiated (IR) VciΔR26 and Vcfl/fl mice and their nonirradiated (N-IR) controls 7 days after irradiation and analyzed using scRNA-seq. (B-C) UMAP plot showing the clustering of integrated nonhematopoietic BM stroma cells from IR VciΔR26 and Vcfl/fl mice and their N-IR controls. UMAP plot showing nonhematopoietic BM stroma cells from irradiated VciΔR26 and Vcfl/fl mice and their N-IR controls separately. Note that damaged ECs (green arrowhead) and Aqp1hi ECs (blue arrowhead) are increased after irradiation. (D) Percentages of the clusters (damaged ECs, Aqp1hi ECs, and Cxcl10 ECs) and quantification of ECs in S phase. (E) Dot plot showing selected differentially expressed genes in SECs (SEC-1, SEC-2, and Cxcl10 ECs), damaged ECs, Aqp1hi ECs, and AECs. Analysis of differentially expressed genes was performed between IR;Vcfl/fl vs N-IR;Vcfl/fl and IR;VciΔR26 vs N-IR;VciΔR26.
Interestingly, 7 days after irradiation, there were more proliferating LepR+ cells (Lepr-1, Vegfahi Lepr-1, and Lepr-2 clusters), which increased further in irradiated VciΔR26 mice (supplemental Figure 8E). The mesenchymal stem cell gene Grem1 and known hematopoietic regenerative transcripts, such as Angpt1, Kitl, Vcam1, and Snail2,6,12,46 were upregulated in LepR+ cells in irradiated Vcfl/fl mice, but not in irradiated VciΔR26 mice (supplemental Figure 8F). In contrast, the expression of S100a8 and S100a9 increased significantly in LepR+ cells in irradiated VciΔR26 mice, but not in irradiated Vcfl/fl mice (supplemental Figure 8F). Taken together, these results suggest that VEGF-C improves hematopoietic niche recovery after irradiation damage by increasing EC proliferation and by enabling a proper regenerative response in both ECs and LepR cells, as suggested by the induction of hematopoietic regenerative genes and suppression of inflammatory molecules.
Discussion
Our work reveals that VEGF-C from BM ECs and LepR+ cells maintains HSC homeostasis by regulating EC-derived signals and LepR+ cells in the perivascular niche. Moreover, VEGF-C promoted vascular and HSC regeneration after myeloablative treatment, implying that VEGF-C contributes to niche repair after injury (Figure 7).
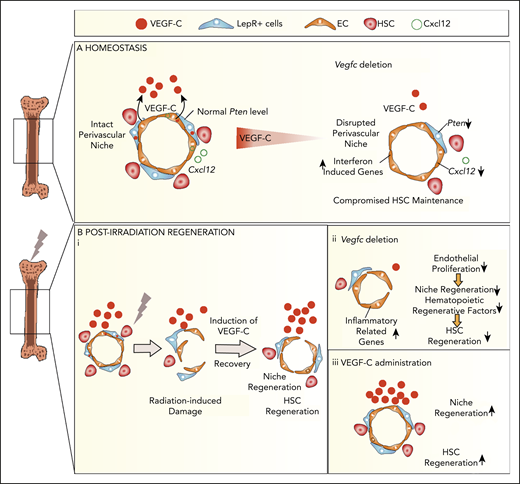
VEGF-C contributes to BM niche maintenance and regeneration after irradiation. Schematic models describing the BM microenvironment of ECs, LepR+ cells, HSCs, and the effects of VEGF-C. (A) During homeostasis, HSCs reside in and are maintained by the intact perivascular niche composed of ECs and LepR+ cells. VEGF-C from both LepR+ cells and ECs maintains an intact BM perivascular niche that is necessary for HSC maintenance. When Vegfc is deleted from ECs or LepR cells, genes related to interferon response are upregulated, and Cxcl12 and Pten are decreased in BM ECs and LepR+ cells, respectively, which leads to niche impairment and compromised HSC maintenance. (Bi) After irradiation and HSC transplantation, Vegfc expression in the niche is upregulated. Microenvironment-derived VEGF-C contributes to endothelial and LepR+ cell regeneration, which in turn is essential for HSC regeneration. (ii) When Vegfc is deleted from the BM microenvironment, genes related to inflammation are upregulated and the proliferation of ECs is decreased, which leads to impaired niche regeneration. Niche-derived hematopoietic regenerative factors are also decreased, resulting in a decreased recovery of HSCs. (iii) Overexpression of VEGF-C improves vascular regeneration, which leads to a better hematopoietic recovery after irradiation or transplantation.
VEGF-C contributes to BM niche maintenance and regeneration after irradiation. Schematic models describing the BM microenvironment of ECs, LepR+ cells, HSCs, and the effects of VEGF-C. (A) During homeostasis, HSCs reside in and are maintained by the intact perivascular niche composed of ECs and LepR+ cells. VEGF-C from both LepR+ cells and ECs maintains an intact BM perivascular niche that is necessary for HSC maintenance. When Vegfc is deleted from ECs or LepR cells, genes related to interferon response are upregulated, and Cxcl12 and Pten are decreased in BM ECs and LepR+ cells, respectively, which leads to niche impairment and compromised HSC maintenance. (Bi) After irradiation and HSC transplantation, Vegfc expression in the niche is upregulated. Microenvironment-derived VEGF-C contributes to endothelial and LepR+ cell regeneration, which in turn is essential for HSC regeneration. (ii) When Vegfc is deleted from the BM microenvironment, genes related to inflammation are upregulated and the proliferation of ECs is decreased, which leads to impaired niche regeneration. Niche-derived hematopoietic regenerative factors are also decreased, resulting in a decreased recovery of HSCs. (iii) Overexpression of VEGF-C improves vascular regeneration, which leads to a better hematopoietic recovery after irradiation or transplantation.
Our findings showed that VEGF-C was produced primarily by LepR+ cells and ECs and that both sources of VEGF-C were functionally required during homeostasis and in response to injury, highlighting the importance of several cell types in coordinating the function of the BM hematopoietic niche. The importance of the paracrine cross-talk between the niche cells is emphasized by our finding that, although the LepR+ cells did not express the known VEGF-C receptors VEGFR-2 or VEGFR-3, the BM ECs acted as cellular intermediates that responded to VEGF-C signaling and consequently promoted the maintenance of BM LepR+ cells. When VEGF-C signaling was disrupted, LepR+ cells were unable to sustain proper levels of Pten, which is so far the only known factor associated with LepR+ cell maintenance.42 VEGF-C signaling also sustained the expression of the key niche factor Cxcl12 (SDF1) in BM ECs. Further studies should identify the specific VEGF-C-induced angiocrine signals from the BM ECs that support the LepR+ cells.
In addition to the endothelium, VEGFR-2 and VEGFR-3 are expressed in BM macrophages47 and megakaryocytes precursors,48 which also support HSC maintenance.49,50 It is not known whether VEGF-C stimulation of these cells influences the hematopoietic niche. Nevertheless, we did not observe significant changes in peripheral blood platelet counts or in the number of myeloid cells in the blood or BM, indicating that loss of VEGF-C did not overtly compromise other niche components besides ECs and LepR+ cells.
Active VEGF-C signaling depends on proteolytic activation of pro-VEGF-C by the complex of pericellular matrix protein CCBE-1 and ADAMTS-3 metalloproteinase.29,51,52 Like VEGF-C, both CCBE-1 and ADAMTS-3 are necessary for embryonic erythropoiesis.53,54 Our scRNA-seq data indicated that the BM LepR+ cells expressed Ccbe1 (data not shown), suggesting its involvement in processing VEGF-C in the adult BM niche.
Our results show that LepR+ cells in the adult BM are decreased upon global or EC-specific deletion of VEGF-C. Although BM ECs were not significantly affected when Vegfc was deleted from adult mice, loss of VEGF-C from LepR+ cells during the early postnatal period compromised endothelial proliferation during a period when the BM vasculature is actively expanding and remodeling. Notably, it has been reported that the Vcfl/fl mice show hypomorphic features in the meningeal lymphatic vessels, even in the absence of Cre.55 Nevertheless, we did not observe defects in the adult BM vasculature in these mice. However, Vegfc deletion caused defective recovery of BM ECs and LepR+ cells after irradiation, leading to compromised hematopoietic regeneration. These results show that genetic deletion of the VEGF-C signaling in actively remodeling vasculature during development or after injury is more harmful than in quiescent adult blood vessels.
In our study, VEGF-C contributed to niche regeneration after irradiation. Analysis of EC transcriptional responses to irradiation by RNA-seq showed that the hematopoietic regenerative genes, such as Angpt2/Tek, Sema3f, and Thbd, were upregulated after irradiation.13,22 Our scRNA-seq analysis also suggests that Vegfc deletion suppresses regenerative EC responses to irradiation, including inhibition of cell proliferation, and induction of the inflammatory molecules S100a8/9. Vegfc deletion also suppressed the expression of hematopoietic regenerative factors (Angpt1, Kitl, Vcam1, and Snail2) in LepR+ cells after irradiation and induced S100a8/9. A previous study showed that inflammatory signaling from the niche involving S100a8/9 induces genotoxic stress in HSPCs and influences the progression of bone marrow failure and myelodysplastic syndrome.56 Furthermore, our data revealed the emergence of damaged ECs and Aqp1hi ECs in the BM after irradiation, which was exaggerated by Vegfc loss. These results together highlight the complexity of the responses of BM ECs and LepR+ cells to irradiation and the critical function for VEGF-C signaling in promoting the regenerative molecular response.
Our current work extends the function of VEGF-C from fetal erythropoiesis21 to the adult BM perivascular niche, BM transplantation, and recovery from irradiation-induced damage. The potential of VEGF-C as a mitigant of irradiation damage in the BM is of great interest, given its ability to protect 2 crucial niche components, ECs and LepR+ cells, as well as HSCs, after radiation-induced injury.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession numbers GSE128464, GSE144420, and GSE153339).
The data that support the findings of this study are available from the corresponding author upon reasonable request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sinem Karaman for critical comments; Riitta Kauppinen and Tapio Tainola for expert technical assistance; the Biomedicum Imaging Unit for microscopy support; the Biomedicum FACS core facility for helping with theh flow cytometry; the AAV gene transfer and cell therapy core facility for the service and providing the AAVs; the FIMM SCA unit for sc-RNA sequencing; and the staff at the Viikki and Biomedicum Helsinki Animal Facilities for excellent animal husbandry.
This work was supported by the Academy of Finland, iCAN-The Digital Precision Cancer Medicine Platform (grant 320185); the Sigrid Jusélius Foundation (K.A.); the Cancer Foundation Finland (K.A.); the Hospital District of Helsinki and a Uusimaa Research Grant; Helsinki Institute of Life Sciences (HiLIFE); the Magnus Ehrnrooth Foundation (S.F.); and the K. Albin Johanssons Foundation (S.F.).
Authorship
Contribution: S.F., S.C., and H.N. performed the experiments; V.-M.L, and M.J. produced and provided VEGF-C protein; D.S. advised on the experimental design and reviewed the manuscript; L.S. contributed to the hypotheses, data interpretation and manuscript writing; and S.F., H.M., and K.A. conceived the project, designed the experiments, analyzed and interpreted the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hanna Mikkola, Department of Molecular, Cell, and Developmental Biology, Eli and Edythe Broad Stem Cell Research Center, University of California, Los Angeles, CA; e-mail: hmikkola@mcdb.ucla.edu; and Kari Alitalo, Biomedicum Helsinki, PL63, Haartmaninkatu 8, University of Helsinki, FIN-00014 Helsinki, Finland; e-mail: kari.alitalo@helsinki.fi.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal