


Abstract
Clonal expansions of mutated hematopoietic cells, termed clonal hematopoiesis, are common in aging humans. One expected consequence of mutation-associated clonal hematopoiesis is an increased risk of hematologic cancers, which has now been shown in several studies. However, the hematopoietic stem cells that acquire these somatic mutations also give rise to mutated immune effector cells, such as monocytes, granulocytes, and lymphocytes. These effector cells can potentially influence many disease states, especially those with a chronic inflammatory component. Indeed, several studies have now shown that clonal hematopoiesis associates with increased risk of atherosclerotic cardiovascular disease. Emerging data also associate clonal hematopoiesis with other nonhematologic diseases. Here, we will review recent studies linking clonal hematopoiesis to altered immune function, inflammation, and nonmalignant diseases of aging.
Introduction
Somatic mutations accumulate over time in all cells.1-5 These mutations are most commonly base substitutions (known as single-nucleotide variants [SNVs]), small insertions or deletions (indels), or copy-number changes of large chromosomal regions (known as structural variants [SVs]). Hematopoietic stem cells (HSCs) are estimated to acquire ∼20 somatic mutations per year in the whole genome6,7 and ∼0.1 mutations per year in protein-coding exons,4 the great majority of which are SNVs. Within the bone marrow, only long-lived HSCs have the capacity for self-renewal for the lifetime of an organism.8 Therefore, in most cases, only mutations arising in HSCs will persist for the lifetime of an individual. Given that there are between ∼50 000 and ∼200 000 HSCs per person,6 it is expected that humans will harbor between 350 000 to 1 400 000 coding mutations within the HSC pool by 70 years of age. If just 1 of these mutations is capable of providing a selective advantage to the HSC in which it arises, clonal expansions in blood should be common during aging.9 Indeed, this phenomenon, termed clonal hematopoiesis, has been strongly linked to aging in several studies of persons unselected for hematologic disorders.10-16
In most studies, the mutations used to define clonal hematopoiesis are similar to those found in hematologic cancers.12-16 The most commonly mutated genes in clonal hematopoiesis include DNMT3A, TET2, ASXL1, JAK2, TP53, and SF3B1 (Figure 1), which are also commonly mutated in acute myeloid leukemia,17,18 myelodysplastic syndrome (MDS),19,20 and myeloproliferative neoplasms (MPNs).21 Consequently, it is not surprising that individuals with clonal hematopoiesis develop these cancers at a higher rate than those without mutations.11,12,22-24 However, the mutations that cause clonal hematopoiesis can also be detected in circulating immune cells such as granulocytes, monocytes, and lymphocytes. This finding raises the possibility that clonal hematopoiesis may result in altered immune responses, which could potentially influence many diseases of aging. Here, we will review recent evidence linking clonal hematopoiesis to nonmalignant diseases of aging.
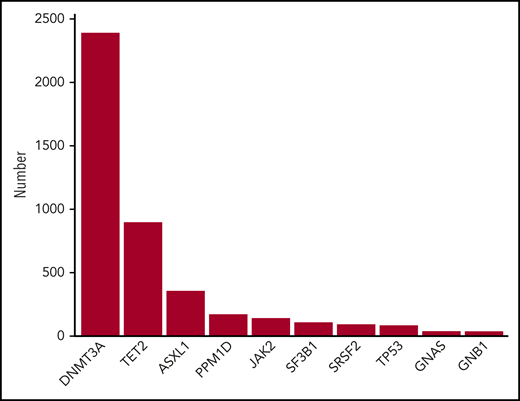
Ten most frequently mutated genes in CHIP. Data were obtained from Bick et al.46
Nomenclature of clonal hematopoiesis
Clonal hematopoiesis refers to any clonal expansion state in the blood-forming system. Blood cancers such as chronic myeloid leukemia or MDS are prototypical examples of clonal hematopoiesis. However, the same mutations found in these cancers are also seen in a large proportion of the healthy elderly population. To distinguish the presence of these mutations in nonmalignant settings from malignant clonal hematopoiesis, the term clonal hematopoiesis of indeterminate potential (CHIP) was introduced.25 CHIP is defined by the presence of a cancer-associated somatic mutation in the blood or bone marrow of persons without a known hematologic cancer or other clonal state, such as monoclonal gammopathy.
Another aspect of the definition of CHIP is a lower bound on the size of the clone. In all studies of clonal hematopoiesis to date, the distribution of clone size is right-tailed; that is, there are many more persons with smaller clones than those with larger clones (Figure 2). Taken to its logical extension, this observation would predict that very small clones should be present in nearly all persons, which has been subsequently demonstrated experimentally.26,27 If these very small clones are present in nearly everyone, then it is no longer a meaningful distinction to be a carrier of clonal hematopoiesis. Because of this fact, we proposed that CHIP must be present at a variant allele fraction (VAF) of >0.02 (meaning >2% of the sequenced alleles bear the mutation, which corresponds to ∼4% of cells for heterozygous mutations). Some commentary has suggested that CHIP is a “technical” definition because this limit is approximately the level of detection from whole-exome sequencing. We prefer to think of CHIP as a sensible working definition: given the aforementioned findings of very small clones in most persons, a threshold VAF is a requirement for the practical design of studies. Although the exact threshold for CHIP is subject to revision, every study to date has shown that the risk of hematologic cancer and other adverse outcomes related to CHIP increases with clone size, and that very small clones (below 0.01-0.02 VAF) have minimal clinical consequence.12,22-24,28-31
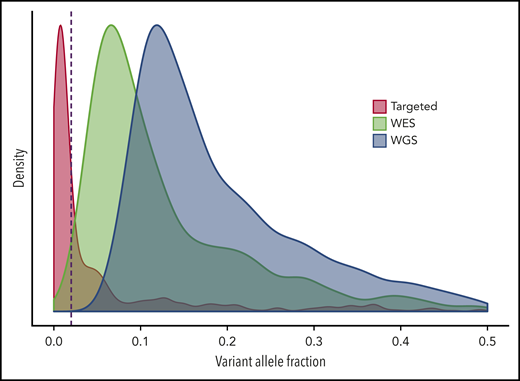
Scaled density plots for VAF of CHIP-associated mutations. Targeted sequencing data were obtained from Abelson et al,22 whole-exome sequencing (WES) data were obtained from Jaiswal et al,12 and whole-genome sequencing (WGS) data were obtained from Bick et al.46 The dashed red line is drawn at variant allele fraction (VAF) 0.02, the cutoff for CHIP. The depth of coverage is the primary factor that determines the shape of the distribution for VAF.
Scaled density plots for VAF of CHIP-associated mutations. Targeted sequencing data were obtained from Abelson et al,22 whole-exome sequencing (WES) data were obtained from Jaiswal et al,12 and whole-genome sequencing (WGS) data were obtained from Bick et al.46 The dashed red line is drawn at variant allele fraction (VAF) 0.02, the cutoff for CHIP. The depth of coverage is the primary factor that determines the shape of the distribution for VAF.
CHIP and immune function
CHIP-associated mutations have been well studied in the context of stem cell biology and malignancy, which has been extensively reviewed elsewhere.32-34 The most common mutations in CHIP are loss-of-function alleles in DNMT3A and TET2, which both encode enzymes involved in DNA methylation.35-38 DNMT3A is the major enzyme responsible for de novo cytosine methylation in hematopoiesis, whereas TET2 is the major enzyme for catalyzing oxidative reactions of methylcytosine bases, which may eventually lead to demethylation. HSCs from mice with null mutations in either of these genes recapitulate the stem cell clonal advantage seen in humans, outcompeting wild-type stem cells in competitive transplant settings.39-42
Recent studies have also uncovered a role for these mutations in immune function, and are best understood in innate immunity. Several studies have found that Tet2-deficient murine macrophages challenged with low-density lipoprotein or bacterial endotoxin more highly express Il1b, Il6, interleukin-8 (IL-8) family chemokines (Cxcl1, Cxcl2, Cxcl3), and other inflammatory mediators compared with wild-type macrophages.28,43,44 Humans with mutations in TET2 are also reported to have increased levels of circulating IL-8,28 IL-6,45 and IL-1B.46 Emerging evidence also supports alterations in innate immune function when DNMT3A perturbed. Mast cells from mice that lack Dnmt3a display increased activity during allergic responses characterized by higher levels on IL-6, tumor necrosis factor α, and IL-13 in response to stimulation with immunoglobulin E.47 Mouse RAW 264.7 macrophages with clustered regularly interspaced short palindromic repeat–induced mutations in Dnmt3a display increased expression of Cxcl1, Cxcl2, and Il6 in response to endotoxin, similar to the phenotype observed in Tet2-deficient macrophages.48 Humans with DNMT3A mutations also display modestly elevated levels of circulating IL-6.46 Mechanistically, very little is known about why these specific gene-expression changes are seen with loss of TET2 or DNMT3A, but may be related to global chromatin changes and enhancer remodeling, as has been observed in HSCs that lack these genes.49
Mutations in JAK2 lead to constitutive signaling from certain growth factor receptors and activation of STAT transcription factors. The result is more robust activation of granulocytes50 and T cells,51 enhanced inflammation in macrophages,52 and activation of neutrophil extracellular traps.53 There is some debate as to whether carrying a JAK2 mutations is de facto proof of MPN, and therefore should not be considered as CHIP.54 However, JAK2 mutations very often occur in persons with normal blood counts, and are found in ∼1 in 1000 middle-aged persons in large population-based surveys, which is a much higher rate than the prevalence of MPNs.12,46,55
Very little is known about how mutations in ASXL1, SF3B1, SRSF2, and other commonly mutated genes affect immune function. Circulating IL-18 has been found to be increased in carriers of SF3B1 mutations,46 whereas circulating IL-6 was increased in carriers of ASXL1 mutations.46 However, despite the myriad proinflammatory molecules associated with CHIP, those who carry these mutations on average do not have increased levels of circulating C-reactive protein.46
It is important to note that the mutations associated with CHIP are nearly always present in circulating granulocytes, monocytes, and natural killer cells, but are only sometimes found in B cells, and rarely in T cells.56 Mutations in DNMT3A are found in the T cells of ∼30% to 50% of DNMT3A-mutated CHIP carriers,56,57 whereas JAK2 mutations are found in the T cells of a majority of patients with JAK2 V617F+ MPNs.51,58,59 In contrast, TET2, ASXL1, or SF3B1 mutations are rarely seen in the T-cell compartment.56 One potential explanation for this finding is that the common CHIP mutations bias differentiation of HSCs away from T-cell lineages, or else arise in progenitors that no longer have potential for T-cell differentiation. An alternative explanation is that T lymphopoiesis has largely ceased by middle age, when a CHIP clone would be reasonably expected to arise.
Despite the lower prevalence of these mutations in the lymphoid compartment, there is emerging evidence that CHIP-associated genes are important in T- and B-cell function. For example, mutations in TET2 and DNMT3A are common in lymphomas of CD4+ T-helper cells, such as angioimmunoblastic T-cell lymphoma.60 There is also a role for these genes in the normal immune function of T cells. Knockout of either Tet2 or Dnmt3a in CD8+ T cells caused a marked expansion of memory T cells in murine models of viral infection.61,62 Interferon γ–producing CD8+ T cells were expanded in both knockouts.
The role of TET2 and DNMT3A in B-cell immunity is less well studied. One group found that loss of Tet2 in B cells of mice led to germinal center hyperplasia upon antigenic stimulation, but impaired affinity maturation and plasma cell formation.63 Similarly, mice that lacked Dnmt3a and Dnmt3b had a higher frequency of activated B cells and expansion of germinal centers upon stimulation.64 Unlike Tet2-deficient mice, these mice had markedly increased plasma cell differentiation and raised antibody titers.
In summary, there is robust evidence to support a role for DNMT3A, TET2, and JAK2 in immune function. Thus, CHIP may be relevant to the phenomenon termed “inflammaging,” the age-associated increase in systemic inflammation.65 In the sections that follow, we will discuss specific associations of these mutations with chronic diseases of aging.
CHIP and atherosclerotic cardiovascular disease
Given that the common CHIP mutations can be found in circulating immune cells and alter their function, it is plausible that the presence of CHIP might influence nonmalignant diseases of aging. This link has been best studied for atherosclerotic cardiovascular disease,66 largely due to the high prevalence of this condition and the availability of large cohorts with detailed clinical phenotypes related to coronary heart disease (CHD) and ischemic stroke.
In 2014, it was reported that CHIP was associated with ∼40% increased mortality, which could not be explained by hematologic cancers alone.12 Instead, CHIP was associated with increased risk of incident CHD and ischemic stroke. The hazard ratio (HR) for CHIP carriers was ∼2 for these diseases, a degree of risk on par with smoking, diabetes, hypertension, and hyperlipidemia.12 These findings were replicated in a large follow-up study that comprised 4 additional case-control cohorts.28 In this study, the presence of CHIP was associated with an approximately twofold increased risk of CHD in older individuals, and an approximately fourfold increased risk of early onset myocardial infarction. Clone size was correlated with the degree of risk, as CHIP carriers with VAF >10% were more likely to develop CHD than those with clones smaller than this size. The HR for CHD was similar in DNMT3A, TET2, and ASXL1 mutation carriers. In contrast, the HR for those with JAK2 V617F mutations was substantially higher.28 The study was not powered to detect associations in less frequently mutated genes in CHIP, like those in spliceosome components (SF3B1, SRSF2, U2AF1) and DNA-damage response (TP53 and PPM1D).
To test causality, 2 groups assessed atherosclerosis in aortae from Ldlr−/− mice that had been transplanted with bone marrow lacking functional alleles of Tet2.28,44 Both groups reported that mice receiving Tet2+/− or Tet2−/− bone marrow developed larger lesions. The same phenotype was observed in mice that lacked Tet2 in only the myeloid compartment, indicating that myeloid dysfunction was sufficient to lead to the phenotype. Tet2−/− mice may develop a progressive monocytosis with age,41 but in the course of these studies, the mice maintained normal blood cell counts and differential. To understand the mechanism, both groups performed gene-expression analyses on macrophages from these mice and found that loss of Tet2 was associated with increased expression of Il1b, Il6, Cxcl1, Cxcl2, Cxcl3, and several other inflammatory molecules, as previously described. Administration of a small molecule inflammasome inhibitor decreased atherosclerotic lesion size to a greater extent in Tet2-mutant mice compared with wild-type mice, suggesting that targeting IL-1B/IL-18 pathways may be beneficial for humans with these mutations.44
Mutations in JAK2 may also result in altered macrophage-inflammatory responses and accelerated atherosclerosis.52 It is unknown whether mutations in SF3B1, SRSF2, and U2AF1 alter atherosclerosis in humans or mice. Some evidence implicates loss of Tp53 in accelerating atherosclerosis in mice,67 but the role of PPM1D mutations in atherosclerosis is unknown. These mutations are often selected after chemotherapy or radiation, so they may be particularly relevant in cancer survivors.68-71
Further evidence for CHIP-associated inflammation promoting atherosclerosis came from analyses of IL6R variants in large human cohorts.29 A common coding variant in IL6R is associated with decreased risk of CHD and lower levels of C-reactive protein.72 Those who carry this protective allele have decreased expression of surface IL-6R on myeloid cells,73 suggesting that activation of this pathway in macrophages may be proatherogenic. In data from the UK Biobank, CHIP due to DNMT3A and TET2 mutations was associated with increased risk of incident CHD and death, similar to prior studies.29 However, carriers of DNMT3A- or TET2-mutated CHIP that had the protective allele for IL6R had a rate of mortality and CHD far below mutation carriers with 2 copies of risk allele (HR, ∼0.5). In contrast, there was no differential effect from this allele in noncarriers of CHIP.29 These data strongly implicate that CHIP causes CHD through a proinflammatory pathway, and that blockade of IL-6R signaling may have clinical benefit specifically in carriers of these mutations.
Given the findings in CHIP carriers, examining cardiovascular phenotypes in families that carry germline loss-of-function alleles in ASXL1,74 TET2,75 or DNMT3A76 will also be of interest. A recent study examined 2 families that had heterozygous carriers of null alleles in TET2.75 Although these carriers never developed myeloid malignancies as would be expected, several unexpectedly developed Hodgkin lymphoma. This suggests that germline mutations in TET2 may have different consequences compared with somatic mutations acquired later in life. Of the 7 carriers evaluated for clinical atherosclerotic disease, 1 had developed CHD by the time of publication, which led the authors to conclude that germline TET2 mutations were likely not causal for cardiovascular disease. However, 4 of the 7 carriers were between ages 19 and 41 years at the time of last contact, and the expected prevalence of CHD in nonmutated persons in this age range would be <1%.77 The other 3 ranged in age from 50 to 63 years, and the expected prevalence of CHD in nonmutated persons is <10% in these age ranges.77 Much larger studies with longer follow-up will be needed to determine whether germline loss of these genes leads to cardiovascular phenotypes.
CHIP and other cardiovascular diseases
Recent work has also found associations between CHIP and other cardiovascular disease states. Those with JAK2-mutated MPNs are well known to have markedly elevated risk of venous and arterial thrombosis.78 However, JAK2-mutated CHIP in the general population was also found to be associated with increased risk of thrombosis.53,55 The exact mechanism of increased thrombosis due to this mutation is still debated, but 1 study found an increased rate of neutrophil extracellular trap formation in JAK2-mutated granulocytes, which is proposed to be a nidus for thrombus formation.53 Surprisingly, mutations in other CHIP-associated genes were also linked to increased risk of venous thrombosis, although to a lower degree than JAK2 mutations.53 The reason for this increased risk is unknown.
Other work has implicated CHIP in the pathogenesis of heart failure (HF). Studies in mice found that mutating Dnmt3a, Tet2, or Jak2 in hematopoietic cells led to worsening cardiac function in murine models of HF.48,79,80 This was attributed to heightened inflammation from monocyte-derived macrophages in the myocardium, and was improved by blocking inflammasome activation. In accordance with the mouse findings, studies in humans with ischemic HF found that CHIP carriers with DNMT3A or TET2 had a markedly increased risk of all-cause mortality.30 Additionally, carriers of these mutations had worsened survival after surgical repair of aortic valve stenosis.81
CHIP was also modestly associated with the presence of type 2 diabetes (T2D) in the same study that first identified a link between CHIP and atherosclerotic cardiovascular disease.12 Another study found that clonal hematopoiesis due to SVs was associated with increased risk of macrovascular complications in those with T2D.82 However, causality in these cases was less certain because T2D anteceded the detection of CHIP, raising the possibility of reverse causation or shared risk factors leading to the observed association. Hyperglycemia has been associated with reduced TET2 enzymatic activity,83 but whether this provides a selective pressure favoring the growth of CHIP clones is unknown. Studies in model systems are needed to further elucidate the nature of the association between CHIP and T2D.
CHIP and noncardiovascular diseases
Relatively little is known about the relationship between CHIP and other nonmalignant diseases. Two groups have reported associations between CHIP and chronic obstructive pulmonary disease,14,84 a disease of aging with a strong inflammatory component. However, both CHIP and chronic obstructive pulmonary disease are also associated with smoking, which may be a confounding factor. Mouse models of lung pathology could be used to understand the cause/effect relationship between CHIP, smoking, and pulmonary disease.
There is epidemiological evidence associating MDS to autoimmune phenomena, which could be explained by acquired stem cell mutations influencing the risk of both diseases.85 One study that looked for an association between CHIP and rheumatoid arthritis found no evidence for such a link.86 However, CHIP-associated mutations are infrequently found in T cells and B cells, which are believed to be the cell types that drive most autoimmune disorders. Studies that look for the presence of mutations in lymphoid cells from patients with these disorders may be warranted. Another study found that ∼30% of persons with antineutrophil cytoplasmic antibody–associated vasculitis harbored a CHIP clone.87 However, controls free of autoimmune disorders were not sequenced, so it is unknown whether CHIP is associated with increased risk of this disorder.
Knowing whether CHIP is associated with age-related neurodegenerative diseases like Alzheimer disease or Parkinson disease would be of great interest, but no studies to date have reported such associations. Brain macrophage populations such as microglia are increasingly implicated in the pathogenesis of these disorders,88 but whether bone marrow–derived macrophages can infiltrate brain parenchyma in normal aging is unknown. One study identified mutations in TET2 and DNMT3A from whole-brain DNA of elderly persons, but the cell type that harbored the mutations was not determined.89
CHIP may also be relevant in the immune response to tumors. Deletion of Dnmt3a in CD8+ T cells prevented T-cell exhaustion and led to enhanced response to programmed cell death protein 1 checkpoint blockade in mice.90 Deletion of Tet2 in murine myeloid cells promoted an antimelanoma T-cell response and reduced tumor size.91 Perhaps more striking are the examples of this phenomenon in clinical settings. Bone marrow transplant recipients who received donor marrow from carriers of DNMT3A-mutated CHIP had reduced relapse rates from their primary tumors and more graft-versus-host disease,92 possibly due to the fact that donor cells that carried the mutations were capable of mounting stronger immune responses against both normal host tissues and the tumor. There is also a report of an exceptional responder with chronic lymphocytic leukemia (CLL) treated by infusion of chimeric antigen receptor (CAR) T cells. The patient harbored TET2-mutant T cells due to CHIP, and the CAR construct fortuitously disrupted the other copy of TET2, leading to a TET2-null CAR-T clone.93 This clone may have been more effective at eliminating CLL because of an expanded central memory CD8+ population, reduced CAR–T-cell exhaustion in response to stimulus, as well as enhanced cytokine production.
A summary of the major reported associations of CHIP is shown in Figure 3.
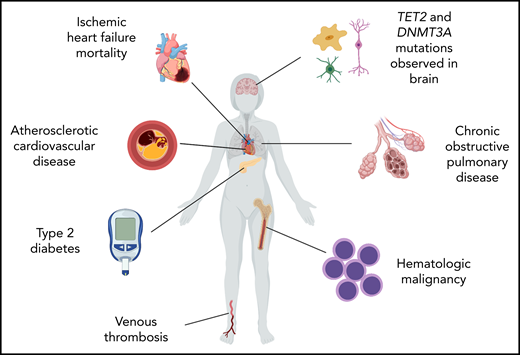
Phenotypic associations of clonal hematopoiesis due to structural variation
The discussion thus far has largely focused on CHIP caused by SNVs and small indels. However, clonal hematopoiesis may also occur due to large SVs such as insertions, deletions, and loss-of-heterozygosity events spanning several kilobases or more.94-96 These large variants are genetically distinct from CHIP due to SNVs or small indels. Although focal deletions or loss-of-heterozygosity events at DNMT3A, TET2, or JAK2 are seen, they are relatively rare. Instead, variants often found in CLL and losses of the sex chromosomes are most common.97 The presence of these large SVs is also associated with an increased risk of hematologic cancer, but the cancers that arise are predominantly CLL.94,95,97 Intriguingly, these variants associate with a doubling of the risk of death, and this effect cannot be explained by deaths due to cancer alone.97 In aggregate, these large SVs do not associate with cardiovascular outcomes, suggesting that the effects seen on atherosclerosis are mutation specific.98
One special case of large SV-associated clonal hematopoiesis occurs due to loss of Y-chromosome (LOY), which is reported to occur in ∼20% of middle-aged men.99 LOY has been associated with smoking,100 mortality,99 cancer,99 cardiovascular outcomes,101 and Alzheimer disease102 in epidemiological studies, but a mechanistic understanding of these associations is lacking. A recent study found that the heritability of LOY estimated from linkage-disequilibrium score regression to be 31.7%,103 compared with 3.6% for CHIP due to SNVs or small indels.46 Application of a polygenic risk score for LOY found that those who carried genetic variants associated with LOY were at higher risk for developing solid cancers.103 This was true even in females who lack a Y-chromosome. The authors speculate that carrying these risk variants might promote underlying genomic instability as a potential explanation for the elevated risk of both hematopoietic LOY in men and solid cancers in women. These results further underscore that all clonal hematopoiesis is not equal, and that causal relationships will need to be established for individual variant types.
Considerations for design of CHIP-phenotype association studies
Upwards of 1 million persons are expected to have sequencing of whole-blood DNA performed as part of large biobank studies in the coming years. Thousands more will be assessed for CHIP in single-center studies. As this field evolves, maintaining rigorous standards for evaluating associations between CHIP and various phenotypes will be critical.
First, investigators should carefully consider the cohorts selected for such studies. Either population-based cohorts unselected for the disease of interest or nested case-control studies from within such cohorts may be used. Ideally, persons free of disease at baseline should be followed longitudinally for the development of disease after the time of DNA sampling for CHIP screening. Time-to-event analyses such as Cox proportional hazards models should be used, and effect sizes should be reported after adjusting for known confounders. The use of retrospective cohorts in which disease occurred prior to the time of DNA sampling is suboptimal due to several biases. First, a temporal relationship between the disease and the CHIP clone cannot be established, which precludes inferences about cause and effect. This study design may also mask a true effect if a large proportion of the prevalent disease cases occurred in the distant past prior to the development of CHIP. Finally, retrospective analyses may introduce survivorship bias; carriers of CHIP may be more likely to die or have morbidity that prevents them from being included in such retrospective studies.
Second, there must be conscientious effort to avoid reporting false-positive or -negative findings. I suggest that investigators perform power calculations to determine whether their study is large enough to robustly detect putative associations before reporting the results of such analyses. Any positive associations in 1 cohort or study should be replicated in an independent cohort, a standard that has been adopted by the broader genetics community. If multiple phenotypes are being assessed in the same study, multiple hypothesis correction should be performed. Lack of statistical significance in a single study does not allow an investigator to conclude that an association does not exist, especially if the study is underpowered for reasonable effect sizes. If these standards are not met, investigators should stress that the findings are merely exploratory and require confirmation.
Third, consistent standards for assays and interpretation of variants must be adopted. CHIP has been detected using whole-genome sequencing, whole-exome sequencing, standard targeted sequencing, and error-corrected targeted sequencing. VAF distribution is highly dependent on the specific sequencing approach (Figure 2), which profoundly affects the observed prevalence of clonal hematopoiesis. Therefore, sequencing cases and controls in a study using the same technologies is critical, and the use of historical controls or comparison with prior published studies is to be avoided. Even when using the same assay, “clonal hematopoiesis” may be defined in several ways by different investigators. CHIP has a very specific definition, so I propose that this definition (or another clearly defined entity) be used as the default for such studies. In most studies, persons were classified as having clonal hematopoiesis only if they carried a cancer-associated somatic variant as reported in the literature or in the Catalogue of Somatic Mutations in Cancer (COSMIC)11,12,16,30,69 ; all other variants should be classified as “variants of unknown significance” and analyzed separately. If numbers permit, gene-specific effects should also be examined. When variants not meeting the definition of CHIP are considered, I suggest that investigators include such variants in separate analyses.
Concluding remarks
CHIP is a prevalent phenomenon in human aging and has a surprisingly broad impact on health and disease. In the coming years, several surprising associations between these mutations and diseases of aging are sure to be revealed. The use of large biobank studies will also provide power to detect factors that influence whether those with CHIP will develop disease. For example, genetic modifiers,29 environmental influences,104 DNA methylation state,105 or other factors may influence the pathogenicity of CHIP. We still have limited understanding of what causes a clone to grow in size in some, but remain stagnant in others. Mutation-specific effects are certain to have a role,106 but what about cell-extrinsic factors? Finally, a greater understanding of the mechanisms by which these mutations alter immune responses to cause disease is needed in order to rationally identify therapies that can be used to prevent the negative consequences associated with CHIP.
Authorship
Contribution: S.J. wrote the paper.
Conflict-of-interest disclosure: S.J. received consulting fees from GRAIL, Inc.
Correspondence: Siddhartha Jaiswal, Stanford University, 240 Pasteur Dr, Room 4654, Stanford, CA 94304; e-mail: sjaiswal@stanford.edu.
