


Abstract
Most arterial thrombotic events have a clear atherosclerotic or cardioembolic etiology, but hematologists are frequently asked to assist in the diagnosis and management of a patient with a nonatherosclerotic and noncardioembolic arterial event, referred to here as an unexplained arterial thrombosis. Because there is an assorted list of factors that can precipitate an arterial event, we present a systematic diagnostic approach to ensure consideration of not only primary hypercoagulable disorders, but also pro-thrombotic medications or substances, vascular and anatomic abnormalities, and undiagnosed systemic disorders, such as malignancy and autoimmune diseases. We also review existing literature of the role of hypercoagulable disorders in arterial thrombosis and discuss our approach to thrombophilia workup in patients after an unexplained arterial event. We conclude with 3 representative cases to both illustrate the application of the outlined diagnostic schema and discuss common management considerations, specifically the selection of anticoagulation vs antiplatelet therapy for secondary prevention.
Introduction
The majority of arterial thromboses are not managed by hematologists. Atherosclerotic plaque rupture, atrial fibrillation, and other cardioembolic sources are responsible for most arterial events, and comprehensive consensus guidelines exist on the evaluation and management of organ-specific arterial thrombotic disorders (Table 1).
Professional society recommendations for antithrombotic therapy for atherosclerotic occlusive arterial disease, atrial fibrillation, valvular heart disease, and patent foramen ovale
However, the approach to nonatherosclerotic and noncardioembolic arterial thrombosis, referred to here as unexplained arterial thrombosis, is less clear. When an arterial event occurs without clear provoking factors, a hematologist is often called upon to address 2 common questions. First, does this patient have an underlying thrombophilia? Second, should this patient be placed on antiplatelet therapy, anticoagulation, or both?
In this article, we first propose a 3-step conceptual framework for the evaluation and management of patients with unexplained arterial thrombosis (Table 2). We then apply this framework to 3 cases to emphasize its utility in clinical management.
Structured approach to the patient referred for unexplained arterial thrombosis
| Step 1: Defining the clot |
|---|
| Review of imaging report |
| Review of imaging studies with radiology |
| Was thrombotic event arterial or venous? |
| Is there evidence of atherosclerosis? |
| Is there a visible vessel wall abnormality? |
| Review of pathology specimens for evidence of atherosclerosis, vasculitis, etc. |
| Discussion of etiology with organ-specific specialist (cardiologist, neurologist, ophthalmologist, etc.) |
| Step 1: Defining the clot |
|---|
| Review of imaging report |
| Review of imaging studies with radiology |
| Was thrombotic event arterial or venous? |
| Is there evidence of atherosclerosis? |
| Is there a visible vessel wall abnormality? |
| Review of pathology specimens for evidence of atherosclerosis, vasculitis, etc. |
| Discussion of etiology with organ-specific specialist (cardiologist, neurologist, ophthalmologist, etc.) |
| Step 2: Performing a diagnostic evaluation (details in Table 3) |
|---|
| A. Is atherosclerosis present or are there atherosclerosis risk factors? |
| B. Has the heart been examined for a cardioembolic source? |
| C. Review of other potential thromboembolic causes |
| a. Medications or substance use |
| b. Systemic diseases |
| c. Vascular or anatomic disorders |
| D. Consideration of thrombophilia evaluation in younger patient without other causative etiology (details in Table 5) |
| Step 2: Performing a diagnostic evaluation (details in Table 3) |
|---|
| A. Is atherosclerosis present or are there atherosclerosis risk factors? |
| B. Has the heart been examined for a cardioembolic source? |
| C. Review of other potential thromboembolic causes |
| a. Medications or substance use |
| b. Systemic diseases |
| c. Vascular or anatomic disorders |
| D. Consideration of thrombophilia evaluation in younger patient without other causative etiology (details in Table 5) |
| Step 3: Determining the management plan |
|---|
| Literature search for up-to-date studies |
| Discussion with organ-specific specialist of best management |
| Consideration of the patient-specific balance between risk of recurrent thrombosis and risk of bleeding |
| Discussion with patient of the limitations of existing data; acknowledgment that antiplatelet and/or anticoagulation treatment decisions are often non-evidence based |
| Reevaluation of patient and antithrombotic therapy on a regular basis |
| Step 3: Determining the management plan |
|---|
| Literature search for up-to-date studies |
| Discussion with organ-specific specialist of best management |
| Consideration of the patient-specific balance between risk of recurrent thrombosis and risk of bleeding |
| Discussion with patient of the limitations of existing data; acknowledgment that antiplatelet and/or anticoagulation treatment decisions are often non-evidence based |
| Reevaluation of patient and antithrombotic therapy on a regular basis |
Conceptual framework for diagnosis and management
Step 1: Defining the clot
The first step in the management of an arterial thromboembolism is to confirm its anatomic location and resultant end-organ damage, often necessitating dedicated review of imaging studies with an expert radiologist. An assessment of the patient’s symptoms as they correlate with the thrombosis location is also helpful. Although some arterial events are incidentally identified, they still require thorough diagnostic evaluation. Discussion with a subspecialist who cares for the affected organ is also recommended because the diagnostic approach to thrombosis in each organ system varies significantly.
For example, when considering thrombosis in the cerebral vasculature, the location of the thrombus or ischemic territory can be suggestive of an embolic vs atherosclerotic etiology. Also, an ischemic cerebral lesion that does not correlate with an anatomical arterial territory, particularly if associated with hemorrhage, raises concern for venous sinus thrombosis, requiring dedicated imaging.12 Splenic and renal infarcts can result from arterial or venous occlusion; wedge-shaped infarcts suggest an arterial origin, whereas diffuse ischemic areas suggest a venous etiology. Retinal vessel thrombosis also requires particular attention, as distinction between retinal artery occlusion and retinal vein occlusion is key, as is central vs branch vessel involvement.13 A discussion with ophthalmology may be necessary to clarify examination findings, including the bilaterality of changes or signs of a local thrombotic or systemic process (vasculitis, sarcoidosis, hyperviscosity, etc).
Defining the vascular obstruction, based on clinical symptomatology, physical examination, imaging studies, and discussion with expert consultants, is an essential first step because thrombosis location and extent of organ damage determine diagnostic considerations and management.
Step 2: Performing a diagnostic evaluation
The potential contributors to an arterial thrombotic event are vast and, therefore, a structured diagnostic evaluation is helpful (Table 3). Because thrombotic events are frequently multifactorial, it is important to identify ALL potential atherosclerotic and thrombotic risk factors.
Suggested approach to structured diagnostic evaluation of unexplained arterial thrombosis
| A. Is atherosclerosis the underlying problem? |
|---|
| Atherosclerotic changes in imaging or pathology specimens? |
| Atherosclerosis risk factors present? |
| Obesity; diabetes mellitus; cigarette smoking; hypertension; high low-density lipoprotein cholesterol; low high-density lipoprotein cholesterol; high lipoprotein(a) |
| Family history of arterial problems in young relatives (<50 y of age) |
| A. Is atherosclerosis the underlying problem? |
|---|
| Atherosclerotic changes in imaging or pathology specimens? |
| Atherosclerosis risk factors present? |
| Obesity; diabetes mellitus; cigarette smoking; hypertension; high low-density lipoprotein cholesterol; low high-density lipoprotein cholesterol; high lipoprotein(a) |
| Family history of arterial problems in young relatives (<50 y of age) |
| B. Has the heart been thoroughly evaluated as an embolic source? |
|---|
| Atrial fibrillation–ECG, extended cardiac rhythm monitor |
| Patent foramen ovale–obtain cardiac echocardiogram: transthoracic with bubble study and Valsalva; if negative, consider transesophageal or transcranial Doppler |
| B. Has the heart been thoroughly evaluated as an embolic source? |
|---|
| Atrial fibrillation–ECG, extended cardiac rhythm monitor |
| Patent foramen ovale–obtain cardiac echocardiogram: transthoracic with bubble study and Valsalva; if negative, consider transesophageal or transcranial Doppler |
| C. Other causes |
|---|
| Is the patient on estrogen therapy (contraceptive pill, ring, or patch; hormone replacement therapy), other hormonal therapy, or prothrombotic cancer therapy? |
| Does the patient use amphetamines, cocaine, or anabolic steroids? |
| Is there evidence of Buerger disease (does patient smoke tobacco or cannabis)? |
| Could the patient have hyperviscosity or cryoglobulins? |
| Is there evidence of a rheumatologic or autoimmune disease? Consider laboratory workup for vasculitis and other immune disorders |
| Were anatomic abnormalities seen in artery leading to the ischemic area (web, fibromuscular dysplasia, dissection, vasculitis, external compression)? |
| Is there a suggestion of an infectious arteritis? |
| Does patient have symptoms of vasospastic disorder (Raynaud)? |
| C. Other causes |
|---|
| Is the patient on estrogen therapy (contraceptive pill, ring, or patch; hormone replacement therapy), other hormonal therapy, or prothrombotic cancer therapy? |
| Does the patient use amphetamines, cocaine, or anabolic steroids? |
| Is there evidence of Buerger disease (does patient smoke tobacco or cannabis)? |
| Could the patient have hyperviscosity or cryoglobulins? |
| Is there evidence of a rheumatologic or autoimmune disease? Consider laboratory workup for vasculitis and other immune disorders |
| Were anatomic abnormalities seen in artery leading to the ischemic area (web, fibromuscular dysplasia, dissection, vasculitis, external compression)? |
| Is there a suggestion of an infectious arteritis? |
| Does patient have symptoms of vasospastic disorder (Raynaud)? |
| D. Thrombophilia workup for arterial events (also see Table 5) |
|---|
| Hemoglobin and platelet count (are cytopenias or cytoses present as evidence of cancer, MPN, or PNH?) |
| Consider the following thrombophilias: |
| FVL, PT20210* |
| PC, PS, AT activities |
| APS evaluation: aCL IgG, IgM; aβ2GPI IgG, IgM; lupus anticoagulant |
| Homocysteine if <30 y of age (to discover homocystinuria) |
| MPN mutation testing if blood count abnormalities present or other evidence for an MPN; consider JAK-2 mutation even if no CBC abnormality present |
| Flow cytometry to assess for PNH if cytopenias or hemolysis present; consider even without such abnormalities |
| Do not test for MTHFR, PAI-1, tPA levels or polymorphisms, FVIII, fibrinogen, or phospholipid antibodies other than those mentioned above |
| D. Thrombophilia workup for arterial events (also see Table 5) |
|---|
| Hemoglobin and platelet count (are cytopenias or cytoses present as evidence of cancer, MPN, or PNH?) |
| Consider the following thrombophilias: |
| FVL, PT20210* |
| PC, PS, AT activities |
| APS evaluation: aCL IgG, IgM; aβ2GPI IgG, IgM; lupus anticoagulant |
| Homocysteine if <30 y of age (to discover homocystinuria) |
| MPN mutation testing if blood count abnormalities present or other evidence for an MPN; consider JAK-2 mutation even if no CBC abnormality present |
| Flow cytometry to assess for PNH if cytopenias or hemolysis present; consider even without such abnormalities |
| Do not test for MTHFR, PAI-1, tPA levels or polymorphisms, FVIII, fibrinogen, or phospholipid antibodies other than those mentioned above |
PAI-1, plasminogen activator inhibitor-1.
Purpose of testing is to discover the homozygous of double heterozygous state (heterozygous FVL plus heterozygous PT20210).
The most common causes of arterial events, atherosclerosis and cardioembolism, must first be excluded (Table 3, section A-B). Evaluation for paroxysmal atrial fibrillation should include electrocardiography (ECG) and ambulatory cardiac rhythm monitoring, the optimal duration of which is debated10,14 ; 2 to 4 weeks is reasonable. Evaluation for patent foramen ovale (PFO) is frequently indicated; because the diagnosis and management of PFO is a complex and evolving field, coevaluation with a knowledgeable cardiologist and neurologist is advisable. Evaluation frequently starts with transthoracic echocardiography (TTE) with an agitated saline study performed while the patient is coughing and/or performing a Valsalva maneuver.15 Transesophageal echocardiography is considered the gold standard for evaluation of PFO, but increasing evidence supports the use of transcranial Doppler as noninvasive option.16
Imaging should be reviewed with a radiologist because diagnostic reports may lack necessary details. Potential questions to discuss include whether (1) the quality of the study is sufficient for vasculature evaluation, (2) there is evidence of atherosclerosis or anatomical abnormalities, and (3) there are features of the damaged organ that clarify arterial vs venous event or embolic vs atherosclerotic etiology.
If no cardioembolic or atherosclerotic explanation is identified, the provider next considers 3 additional categories of conditions that increase arterial thrombotic risk: medications or substance use, systemic diseases, and vascular/anatomic disorders (Table 3, section C).
Medications or substance use
A thorough review of a patient’s medications is necessary because combined oral contraceptives (COC),17 hormone replacement therapy,18 anabolic androgenic steroid use,19 and intravenous immunoglobulin20 may variably increase arterial thrombotic risk. Multiple anticancer agents also increase risk, including inhibitors of vascular endothelial growth factor (ie, bevacizumab, sorafenib) and l-asparaginase.21
Estrogen-containing medications are most well-studied, with comprehensive reviews of the associated increased cardiovascular and stroke risk previously published.22,23 The risk of venous thrombosis has been shown to be highest within the first months of COC initiation, but the generalizability of these findings to arterial thrombosis is unclear.24 Management considerations for patients with arterial events while taking COCs are addressed in case 3. The association between testosterone supplementation and arterial thromboembolism is less clear, but there is concern and controversy regarding its potential to increase risk of cardiovascular events.25,26
Heparin-induced thrombocytopenia should be considered in any hospitalized patient because heparin exposure can occur through undocumented heparin flushes, and platelet count can decline without reaching a classically “low” threshold.
A thorough history of substance use and urine drug screen is warranted. Cocaine has multiple acute and long-term pro-thrombotic effects.27 Both tobacco, including smokeless tobacco,28 and marijuana29 can precipitate thromboangiitis obliterans (Buerger disease), which may manifest with arterial thrombosis.
Systemic diseases
Many systemic disorders may first present with an arterial event.
In patients with cancer, the rate of arterial thrombosis is 4.7% in the 6 months after cancer diagnosis, attributable to both active malignancy and pro-thrombotic treatments.30 In patients without a cancer diagnosis, age-appropriate cancer screening should be performed because patients have an increased arterial thrombotic risk before cancer diagnosis.31-33 The value of more extensive malignancy evaluation after an arterial thromboembolism has not been studied, but data in venous thromboembolism argue against it.34
Myeloproliferative neoplasms (MPN) and paroxysmal nocturnal hemoglobinuria (PNH) are associated with significantly increased thrombotic risk, particularly arterial events.35,36 Appropriate testing should be pursued in patients with blood count abnormalities (cytopenias, cytoses) or evidence of hemolysis. Whether there is benefit to testing for JAK2V617F or PNH in the absence of hematologic findings is unclear given limited existing data.37-39
Hyperviscosity, whether from erythrocytosis, leukocytosis, or excess plasma proteins (plasma cell disorder, cryoglobulinemia), can also increase arterial thrombotic risk. Sickle cell disease is more commonly associated with stroke versus other arterial events. Hemoglobin SC and S-β-thalassemia may have a benign clinical course before thromboembolism.40
There is a well-documented correlation between systemic inflammatory disorders and increased arterial thrombotic risk,41,42 including sarcoidosis43 and systemic vasculitides such as antineutrophil cytoplasmic antibodies-associated vasculitis, large-vessel vasculitis, and Behçet’s syndrome.44 Therefore, a thorough evaluation for signs and symptoms of autoimmune disorders is warranted; nonspecific symptoms include generalized fatigue, arthralgias, fever, rash, and neuropathy, whereas more specific signs include palpable purpura, bruits, blood pressure discrepancies, or combined renal and pulmonary involvement. Imaging can be reviewed with an expert radiologist for findings of vasculitis.45 Laboratory evaluation could include urinalysis and inflammatory markers (erythrocyte sedimentation rate, C-reactive protein). If suspicion is high, disease-specific markers can be obtained (Figure 1) and referral to rheumatology considered.
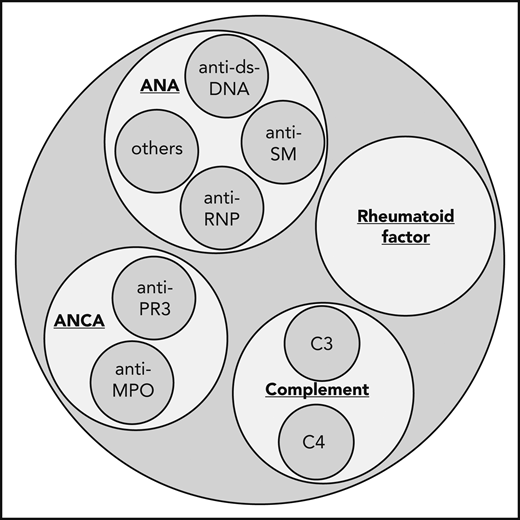
Which antibodies to test for in the evaluation of vasculitis. When considering workup for an autoimmune and vasculitic etiology of an unexplained arterial thromboembolism, consider ordering erythrocyte sedimentation rate, C-reactive protein, ANA, rheumatoid factor, ANCA, and C3 and C4 complement. ANA, antinuclear antibodies; ANCA, antineutrophil cytoplasmic antibodies; ds-DNA, double-stranded DNA; MPO, myeloperoxidase; PR3, proteinase 3; RNP, ribonucleoprotein; SM, SM proteins (core proteins of small nuclear ribonucleoproteins).
Which antibodies to test for in the evaluation of vasculitis. When considering workup for an autoimmune and vasculitic etiology of an unexplained arterial thromboembolism, consider ordering erythrocyte sedimentation rate, C-reactive protein, ANA, rheumatoid factor, ANCA, and C3 and C4 complement. ANA, antinuclear antibodies; ANCA, antineutrophil cytoplasmic antibodies; ds-DNA, double-stranded DNA; MPO, myeloperoxidase; PR3, proteinase 3; RNP, ribonucleoprotein; SM, SM proteins (core proteins of small nuclear ribonucleoproteins).
Vascular or anatomic disorders
Abnormalities in the vessel wall include dissection and vasculitis, as well as less common disorders, such as fibromuscular dysplasia.46 Segmental arterial mediolysis47 and vascular Ehlers-Danlos syndrome48 more commonly present with arterial rupture and hemorrhage, but can also precipitate thrombosis. Certain disorders characteristically occur in a given arterial location, with cystic adventitial disease occurring in the popliteal artery and endofibrosis in the external iliac arteries.49 Extrinsic arterial compression, as in popliteal artery entrapment syndrome and thoracic outlet syndrome,50 can also occur. Vasospasm, idiopathic or secondary to autoimmune disease or substances (eg, cocaine, amphetamines, β-blockers, certain chemotherapy and migraine medications),51 can cause transient ischemia and long-term vascular remodeling that increase thrombotic risk. This classically occurs in the peripheral extremities, as in Raynaud phenomenon,52 but can also involve coronary, cerebral, and mesenteric arteries.53,54 Peripheral organ infarction (ie, kidney, spleen) can be the result of thromboembolism, which may arise from the aorta.55 Furthermore, it can be caused by vessel wall abnormalities such as dissection, aneurysm, and rare disorders including fibromuscular dysplasia and segmental arterial mediolysis.
For each of these diagnoses, dedicated imaging, often with traditional contrast angiography, is required to look for vascular stenosis, dilation, occlusion, compression, aneurysm, or other characteristic findings.
Considering thrombophilia testing
The role of thrombophilias in arterial thrombosis is not well-defined, as opposed to venous thrombosis in which thrombophilias have been more extensively studied.56 The antiphospholipid syndrome (APS), however, has a well-documented increased venous and arterial thrombotic risk and is specifically addressed later.
Table 4 lists seminal publications of the role of thrombophilias in arterial thrombosis. As a whole, these studies have important limitations. First, the majority are retrospective; few prospective randomized trials exist to evaluate the clinical significance or preferred management strategy of thrombophilias in arterial disease. Second, the ability to identify or exclude a correlation is limited by low prevalence of both thrombophilias and truly unexplained arterial thrombosis. Third, the majority of studies quantify the risk associated with an index thrombosis, but the risk associated with recurrence is unknown.
Seminal publications on the association between commonly tested thrombophilias and arterial thrombotic events
| Disorder | Reference | Outcome | Age or other characteristic | Genotype or diagnostic category | Result (95% CI) |
|---|---|---|---|---|---|
| FVL | Kim et al58 | MI, stroke, PVD | All ages | Heterozygous* | OR 1.21 (0.99-1.49) |
| <55 y | OR 1.37 (0.96-1.97) | ||||
| Ye et al59 | MI, CAD | NS | Per-allele RR 1.17 (1.08-1.28) | ||
| Mannucci et al60 | MI | <45 y | OR 1.66 (1.15-2.38) | ||
| Chiasakul et al63 | Stroke | All ages | OR 1.23 (1.05-1.45) | ||
| Homozygous | OR 2.24 (1.26-4.71) | ||||
| PT20210 | Kim et al58 | MI, stroke | All ages | Heterozygous* | OR 1.32 (1.03-1.69) |
| <55 y | OR 1.66 (1.13-2.46) | ||||
| Ye et al59 | MI, CAD | NS | Per-allele RR 1.31 (1.12-1.52) | ||
| Mannucci et al60 | MI | <45 y | OR 1.28 (0.91-1.79) | ||
| Vazquez et al61 | PVD | NS | OR 1.68 (0.8-3.2) | ||
| CLI | OR 3.2 (1.6-6.1) | ||||
| Chiasakul et al63 | Stroke | All ages | OR 1.41 (1.13-1.76) | ||
| Homozygous | OR 7.19 (2.47-20.94) | ||||
| PC | Mahmoodi et al64 | MI, stroke, TIA, PVD | >15 y | NS | OR 6.9 (2.1-22.2) |
| Chiasakul et al63 | Stroke | All ages | OR 2.13 (1.16-3.90) | ||
| PS | Mahmoodi et al64 | MI, stroke, TIA, PVD | >15 y | NS | OR 4.6 (1.1-18.3) |
| Chiasakul et al63 | Stroke | All ages | OR 2.26 (1.34-3.80) | ||
| AT | Mahmoodi et al64 | MI, stroke, TIA, PVD | >15 y | NS | OR 1.1 (0.1-10.9) |
| Chiasakul et al63 | Stroke | All ages | 1.25 (0.58-2.67) | ||
| APS | Neville et al68 | MI, angina,stroke, TIA, other | >18y | Number ab present (per 1-ab difference) | OR 1.46 (0.93-2.27) |
| aCL + LAC + aß2GPI | OR 3.20 (0.60-17.18) | ||||
| FVIII | Zakai et al73 | Stroke | ≥45 y | Per SD increase | HR 1.26 (1.08-1.46) |
| CAD | Per SD increase | HR 1.52 (1.29-1.79) | |||
| Folsom et al74 | CAD | 45-84 y | Highest quartile elevation | HR 1.13 (0.80-1.75) | |
| Homocysteine | Homocysteine Studies Collab76 | CAD | NS | Highest quintile elevation | OR 1.16 (1.02-1.32) |
| MTHFR | Kim et al58 | MI, stroke | All ages | Homozygous C677T | OR 1.20 (1.02-1.41) |
| <55 y | OR 1.41 (1.13-1.76) | ||||
| Klerk et al80 | CAD | European | OR 1.14 (1.01-1.28) | ||
| North American | OR 0.87 (0.73-1.05) |
| Disorder | Reference | Outcome | Age or other characteristic | Genotype or diagnostic category | Result (95% CI) |
|---|---|---|---|---|---|
| FVL | Kim et al58 | MI, stroke, PVD | All ages | Heterozygous* | OR 1.21 (0.99-1.49) |
| <55 y | OR 1.37 (0.96-1.97) | ||||
| Ye et al59 | MI, CAD | NS | Per-allele RR 1.17 (1.08-1.28) | ||
| Mannucci et al60 | MI | <45 y | OR 1.66 (1.15-2.38) | ||
| Chiasakul et al63 | Stroke | All ages | OR 1.23 (1.05-1.45) | ||
| Homozygous | OR 2.24 (1.26-4.71) | ||||
| PT20210 | Kim et al58 | MI, stroke | All ages | Heterozygous* | OR 1.32 (1.03-1.69) |
| <55 y | OR 1.66 (1.13-2.46) | ||||
| Ye et al59 | MI, CAD | NS | Per-allele RR 1.31 (1.12-1.52) | ||
| Mannucci et al60 | MI | <45 y | OR 1.28 (0.91-1.79) | ||
| Vazquez et al61 | PVD | NS | OR 1.68 (0.8-3.2) | ||
| CLI | OR 3.2 (1.6-6.1) | ||||
| Chiasakul et al63 | Stroke | All ages | OR 1.41 (1.13-1.76) | ||
| Homozygous | OR 7.19 (2.47-20.94) | ||||
| PC | Mahmoodi et al64 | MI, stroke, TIA, PVD | >15 y | NS | OR 6.9 (2.1-22.2) |
| Chiasakul et al63 | Stroke | All ages | OR 2.13 (1.16-3.90) | ||
| PS | Mahmoodi et al64 | MI, stroke, TIA, PVD | >15 y | NS | OR 4.6 (1.1-18.3) |
| Chiasakul et al63 | Stroke | All ages | OR 2.26 (1.34-3.80) | ||
| AT | Mahmoodi et al64 | MI, stroke, TIA, PVD | >15 y | NS | OR 1.1 (0.1-10.9) |
| Chiasakul et al63 | Stroke | All ages | 1.25 (0.58-2.67) | ||
| APS | Neville et al68 | MI, angina,stroke, TIA, other | >18y | Number ab present (per 1-ab difference) | OR 1.46 (0.93-2.27) |
| aCL + LAC + aß2GPI | OR 3.20 (0.60-17.18) | ||||
| FVIII | Zakai et al73 | Stroke | ≥45 y | Per SD increase | HR 1.26 (1.08-1.46) |
| CAD | Per SD increase | HR 1.52 (1.29-1.79) | |||
| Folsom et al74 | CAD | 45-84 y | Highest quartile elevation | HR 1.13 (0.80-1.75) | |
| Homocysteine | Homocysteine Studies Collab76 | CAD | NS | Highest quintile elevation | OR 1.16 (1.02-1.32) |
| MTHFR | Kim et al58 | MI, stroke | All ages | Homozygous C677T | OR 1.20 (1.02-1.41) |
| <55 y | OR 1.41 (1.13-1.76) | ||||
| Klerk et al80 | CAD | European | OR 1.14 (1.01-1.28) | ||
| North American | OR 0.87 (0.73-1.05) |
Note: Studies devoted to pediatric populations (<18 y) not included.
ab, antibody; CAD, coronary artery disease; CLI, critical limb ischemia; LA, lupus anticoagulant; MI, myocardial infarction; NS, not specified; PVD, peripheral vascular disease; SD, standard deviation; TIA, transient ischemic attack.
Most cases are heterozygous but presumably there are a few homozygous included, with exact numbers not reported.
Assessing the results in Table 4 as a whole, the association between a given thrombophilia and arterial thrombosis is generally weak. The potential danger of testing, therefore, arises from attributing complete causality to an identified thrombophilia. For example, if a laboratory abnormality suggests a thrombophilia, a patient may be inappropriately placed on lifelong anticoagulation without further workup, posing significant risk to the patient without proven benefit. Alternatively, failure to identify a clinically significant thrombophilia could lead to inappropriate or inadequate treatment.
With these important caveats, we review existing data and present our approach to testing in patients with unexplained arterial thrombosis. Our approach to the management of an identified thrombophilia (ie, the use of anticoagulation, antiplatelet therapy, or both for secondary prevention) is discussed in case 1 and summarized in Table 5. Notably, the studies reviewed include predominantly adult patients; studies in children (<18 years of age) indicate a stronger association between an incident stroke event, but data on recurrence risk are lacking.57
Summary of evidence for thrombophilia testing practices and considerations for anticoagulation vs antiplatelet therapy
| Thrombophilia | Summary of evidence | Testing | Anticoagulation vs antiplatelet | |
|---|---|---|---|---|
| FVL | Heterozygous | Evidence against association with MI, CAD, PVD in all-comers. Small association with stroke in all-comers and MI in patients <45-55 y; clinical significance unclear | Consider testing to identify homozygous FVL or double heterozygous FVL/PT | No influence |
| Homozygous | Insufficient data to clearly identify association with arterial thrombosis | Anticoagulation and/or antiplatelet therapy could be considered | ||
| PT20210 | Heterozygous | Small association with MI, CAD, stroke; clinical significance unclear. Evidence against association with PVD. | Consider testing to identify homozygous PT or double heterozygous FVL/PT | No influence |
| Homozygous | Insufficient data to clearly identify association with arterial thrombosis | Anticoagulation and/or antiplatelet therapy could be considered | ||
| PC | Moderate association with MI, stroke, TIA, PVD in younger patients (<55 y) | Consider testing in patients <55 y | Anticoagulation and/or antiplatelet therapy could be considered | |
| PS | ||||
| AT | Insufficient data to identify association with arterial thrombosis | Consider testing in patients <55 y. Testing based on expert guidelines66 | Anticoagulation and/or antiplatelet therapy could be considered | |
| APS | Proven association with arterial thrombosis | Recommended in patients with no etiology identified. Testing based on expert guidelines67 | Some experts favor anticoagulation; antiplatelet and/or anticoagulation could be considered; initial data suggest DOACs inferior to warfarin | |
| FVIII | Inconsistent correlation with arterial thrombosis | Not recommended | No influence | |
| Homocysteine | Slight association with CAD, stroke; however, no benefit of therapy to lower levels | Consider testing only in patients <30 y if concern for homocystinuria | No influence | |
| MTHFR* | No consistent association with arterial thrombosis | Not recommended | No influence | |
| Thrombophilia | Summary of evidence | Testing | Anticoagulation vs antiplatelet | |
|---|---|---|---|---|
| FVL | Heterozygous | Evidence against association with MI, CAD, PVD in all-comers. Small association with stroke in all-comers and MI in patients <45-55 y; clinical significance unclear | Consider testing to identify homozygous FVL or double heterozygous FVL/PT | No influence |
| Homozygous | Insufficient data to clearly identify association with arterial thrombosis | Anticoagulation and/or antiplatelet therapy could be considered | ||
| PT20210 | Heterozygous | Small association with MI, CAD, stroke; clinical significance unclear. Evidence against association with PVD. | Consider testing to identify homozygous PT or double heterozygous FVL/PT | No influence |
| Homozygous | Insufficient data to clearly identify association with arterial thrombosis | Anticoagulation and/or antiplatelet therapy could be considered | ||
| PC | Moderate association with MI, stroke, TIA, PVD in younger patients (<55 y) | Consider testing in patients <55 y | Anticoagulation and/or antiplatelet therapy could be considered | |
| PS | ||||
| AT | Insufficient data to identify association with arterial thrombosis | Consider testing in patients <55 y. Testing based on expert guidelines66 | Anticoagulation and/or antiplatelet therapy could be considered | |
| APS | Proven association with arterial thrombosis | Recommended in patients with no etiology identified. Testing based on expert guidelines67 | Some experts favor anticoagulation; antiplatelet and/or anticoagulation could be considered; initial data suggest DOACs inferior to warfarin | |
| FVIII | Inconsistent correlation with arterial thrombosis | Not recommended | No influence | |
| Homocysteine | Slight association with CAD, stroke; however, no benefit of therapy to lower levels | Consider testing only in patients <30 y if concern for homocystinuria | No influence | |
| MTHFR* | No consistent association with arterial thrombosis | Not recommended | No influence | |
CAD, coronary artery disease; DOAC, direct oral anticoagulant; MI, myocardial infarction; PVD, peripheral vascular disease; TIA, transient ischemic attack.
MTHFR polymorphisms are not considered to be a thrombophilia.
Factor V Leiden or prothrombin 20210 mutation
Multiple studies of factor V Leiden (FVL) and prothrombin 20210 mutation (PT20210) have illustrated a small association between the heterozygous state with various sites of arterial thrombosis (Table 4).58-61 There has been less investigation into homozygosity or double heterozygosity (FVL and PT20210); 1 retrospective family cohort revealed that collectively, these patients had a nonsignificant 1.6-fold (95% confidence interval [CI], 0.7-3.9) increased risk of cardiovascular disease compared with heterozygous patients,62 whereas a large meta-analysis indicates a significantly increased risk of stroke (FVL: odds ratio [OR], 2.24; 95% CI, 1.26-4.71; PT20210: OR, 7.19; 95% CI, 2.47-20.94).63
The clinical significance of the risk associated with heterozygosity is unclear, but given the small degree of it and the current lack of evidence on how heterozygosity may influence management, we do not test patients to identify heterozygous states. However, given that existing data in homozygosity, limited as they are, suggest a more substantial arterial thrombosis risk, we do consider testing with the intent to identify homozygous or double heterozygous states, acknowledging a similar lack of evidence to guide how homozygosity influences management.
Protein C, protein S, or antithrombin deficiency
The role of deficiencies of protein C (PC), protein S (PS), and antithrombin (AT) in arterial thromboembolism was evaluated most comprehensively in a retrospective family study.64 When compared with family members without thrombophilia, the risk of first arterial event increased by 4.6-fold with PS deficiency (95% CI, 1.1-18.3) and 6.9-fold with PC deficiency (95% CI, 2.1-22.2), but did not in AT deficiency (OR, 1.1; 95% CI, 0.1-10.9). The lack of association with AT deficiency may seem surprising, but the study did not report whether families had less prothrombotic AT deficiency because of a heparin binding defect, or the more prothrombotic type I, IIA, and IIC deficiencies.65
Given the association between PC and PS deficiency and limited data on AT deficiency and arterial thrombosis, we consider testing for these 3 deficiencies in patients younger than 55 years of age (with AT testing performed per guidelines to identify prothrombotic subtypes).66
Antiphospholipid syndrome
APS is well-known to increase risk for arterial thromboembolic events. Diagnosis is based on the updated Sapporo Criteria (Sydney Criteria), requiring a history of thrombosis and/or pregnancy morbidity as well as laboratory evidence of antiphospholipid antibodies (APLA), persistent on 2 samples 12 weeks apart, as measured by 3 assays: lupus anticoagulant (LA), anticardiolipin (aCL) IgG and IgM antibodies, and anti-β2-glycoprotein-I (aβ2GPI) IgG and IgM antibodies.67 The risk of thrombotic events in APS correlates with the number of positive assays, although correlation is more significant with venous than arterial events (with 1 representative study presented in Table 4).68
APS should be considered in all patients with unexplained arterial thrombosis. However, testing has caveats: the LA assay may be falsely abnormal in patients receiving anticoagulants, including warfarin, heparins, and the direct oral anticoagulants (although some assays contain heparin neutralizer to minimize false positives). Antibody titers can be transiently elevated in the setting of acute inflammation. Testing for aCL and aβ2GPI IgA is controversial and testing for antibodies against other phospholipids or phospholipids binding proteins is not supported by current data.67,69,70 Also, emerging data question the risk associated with isolated aCL and/or aß2GPI IgM elevation.71
Unique management considerations in APS are discussed in case 2.
Factor VIII elevation
Studies investigating the role of factor VIII (FVIII) in arterial thrombosis have found conflicting results. A thorough review previously concluded that, although studies have shown an association between high FVIII levels and thrombotic risk, the risk increase is lower than that of classical risk factors, there is significant result variability because of patient-specific and laboratory testing parameters, and, therefore, levels have inconsistent therapeutic implications.72 Additional studies since that time have not significantly changed these conclusions (Table 4).73,74
Given the inconsistency of study results and the variability of FVIII level and testing strategy, we do not routinely test FVIII levels.
Elevated homocysteine, methylenetetrahydrofolate reductase polymorphisms
Homocystinuria is a genetic metabolic disorder leading to very high serum homocysteine levels (typically >100 μmol/L), a high risk of arterial thromboembolism, and characteristic manifestations (Marfanoid habitus, nearsightedness, dislocated lens, intellectual disability) in children and young adults.75 Homocysteinemia, on the other hand, refers to mild or moderately elevated serum homocysteine and its association with atherosclerosis and arterial thrombosis is small and of questionable significance.76 Furthermore, therapy to lower homocysteine levels in homocysteinemia has not been consistently shown to decrease thrombotic risk.77,78
Methylenetetrahydrofolate reductase (MTHFR) plays a key role in folate metabolism and may cause homocysteinemia. MTHFR polymorphisms, most commonly C to T substitution at nucleotide 677, are exceedingly prevalent79 and have not been consistently associated with arterial thrombotic risk.58,80
Given the lack of association between homocysteinemia, MTHFR polymorphisms, and arterial events, we recommend against testing. An exception is young patients (<30 years) in whom there is concern for homocystinuria driven by other characteristic manifestations.
Other
Many other factors have been considered in the search for contributors to arterial thromboembolism, particularly abnormalities in the fibrinolytic pathway (fibrinogen level and polymorphisms), plasminogen deficiency, increased tissue plasminogen activator level and polymorphism, plasminogen activator inhibitor-1 level and 4G/5G polymorphism, and thrombin-activatable fibrinolysis inhibitor levels.81-85 None have shown a consistent association with arterial thromboembolism as reviewed elsewhere,86 and therefore routine testing is not advised.
Step 3: Determining the management plan
The management of an unexplained arterial thrombosis is challenging to generalize because it is case- and patient-specific. Therefore, in the following 3 cases, we illustrate the application of our diagnostic framework and discuss management, both specific to the case and also as generalizable principles for clinical practice.
Cases
Case 1: 39-year-old man with acute stroke
A 39-year-old previously healthy man presented with sudden onset aphasia and right-sided weakness. Magnetic resonance imaging (MRI) scans revealed an acute infarct in the left middle cerebral artery distribution. Deficits resolved following tissue plasminogen activator (tPA). Basic laboratory testing was normal. He denied substance use, and urine drug screen was negative. He had no family members with stroke or other thrombotic disorders. Blood pressure and screening for atherosclerotic risk factors (lipid panel, lipoprotein(a), hemoglobin A1c) were normal. ECG and cardiac monitor during 48-hour hospitalization were without arrhythmia, as was a 30-day Holter monitor after discharge. TTE with bubble study with Valsalva showed no evidence of right-to-left shunt, valvular disease, or intracardiac thrombus. Bilateral duplex carotid ultrasounds and computed tomography (CT) angiography of head and neck were without abnormalities. He was discharged from the hospital on aspirin. Given no clear etiology of his stroke, the patient presented to hematology clinic 1 month after discharge for concern for thrombophilia.
Approach to case 1.
This patient had a medium-sized vessel arterial thrombosis without identified cause. History and initial workup revealed no obvious atherosclerotic or cardioembolic etiology. No causative medications or other substances were identified and there were no symptoms or signs of a systemic disorder with normal physical exam (including a testicular exam), complete blood cell count (CBC), and liver enzymes. Imaging was reviewed with radiology and there was no evidence of anatomical abnormalities in the cerebral circulation.
In any patient with thromboembolism, it is appropriate to consider occult malignancy (see “Systemic diseases”). If malignancy is identified, uncertainty exists whether antiplatelet vs anticoagulation therapy should be used for secondary prophylaxis because high-quality data are lacking. The stroke literature has traditionally favored anticoagulation,87 although recent data suggest aspirin may have similar efficacy to anticoagulation with rivaroxaban, with less bleeding risk.88 This patient’s young age, absence of clinical signs or symptoms, and absence of family history made malignancy unlikely. In the absence of indications for age-appropriate screening, no additional workup for malignancy was pursued.
Strokes without identifiable causative etiology are referred to as cryptogenic, but a new term, embolic stroke of undetermined source (ESUS), was coined in 2014 to address a subgroup of cryptogenic strokes that appear thromboembolic despite inability to identify an embolic source.89 This patient meets criteria for ESUS as outlined in Table 6. It was hypothesized that patients with ESUS would benefit from secondary prevention with anticoagulation rather than antiplatelet therapy; however, recent randomized comparisons of rivaroxaban90 and dabigatran91 to aspirin revealed no improvement in secondary stroke prevention and increased bleeding risk with anticoagulants. Because the designation does not currently change management, some neurologists argue the concept of ESUS is not clinically useful, although further refinement of the definition92 and studies of other anticoagulants93 may ultimately influence clinical practice.
Criteria for diagnosis of embolic stroke of undetermined source89
| Four diagnostic criteria |
|---|
| 1. Nonlacunar ischemic stroke on CT or MRI |
| “Lacunar” defined as subcortical infarct ≤1.5 cm (≤2.0 cm on MRI diffusion images) in largest dimension, including on MRI diffusion-weighted images, and in distribution of small, penetrating cerebral arteries of cerebral hemispheres and pons; visualization by CT usually needs delayed imaging >24-28 h after stroke onset |
| 2. Absence of atherosclerosis (extra- or intracranial) causing ≥50% luminal stenosis in arteries supplying the ischemic area |
| 3. No major risk cardioembolic source |
| “Major risk” source includes atrial fibrillation (permanent or paroxysmal), sustained atrial flutter, intracardiac thrombus, prosthetic cardiac valve, cardiac tumors, mitral stenosis, recent (<4 wk) myocardial infarction, left ventricular ejection fraction <30%, valvular vegetations, or infective endocarditis |
| 4. No other specific cause of stroke identified |
| Other causes including arteritis, dissection, vasospasm, substance use |
| Four diagnostic criteria |
|---|
| 1. Nonlacunar ischemic stroke on CT or MRI |
| “Lacunar” defined as subcortical infarct ≤1.5 cm (≤2.0 cm on MRI diffusion images) in largest dimension, including on MRI diffusion-weighted images, and in distribution of small, penetrating cerebral arteries of cerebral hemispheres and pons; visualization by CT usually needs delayed imaging >24-28 h after stroke onset |
| 2. Absence of atherosclerosis (extra- or intracranial) causing ≥50% luminal stenosis in arteries supplying the ischemic area |
| 3. No major risk cardioembolic source |
| “Major risk” source includes atrial fibrillation (permanent or paroxysmal), sustained atrial flutter, intracardiac thrombus, prosthetic cardiac valve, cardiac tumors, mitral stenosis, recent (<4 wk) myocardial infarction, left ventricular ejection fraction <30%, valvular vegetations, or infective endocarditis |
| 4. No other specific cause of stroke identified |
| Other causes including arteritis, dissection, vasospasm, substance use |
Given the absence of a thromboembolic etiology having considered Table 3 sections A-C, the value of thrombophilia testing (Table 3, section D) was discussed with the patient, highlighting that the intention of testing would be to identify a thrombophilia that may lead to the use anticoagulation ± aspirin for secondary prevention rather than aspirin alone. Our approach to thrombophilia testing and its role in agent selection is outlined in Table 5. Importantly, there is no evidence to support the superiority of anticoagulation, antiplatelet therapy, or the combination. Therefore, as always with non-evidence-based antithrombotic management decisions, incorporation of patient-specific factors including bleeding risk, site of thrombosis, and patient preference, are of high importance. Finding heterozygous FVL or PT20210 alone would not influence our management decision given the small association of questionable clinical significance with arterial thrombotic risk (see “Factor V Leiden or Prothrombin 20210 Mutation”) and aspirin would be our treatment of choice. However, for secondary prevention in patients homozygous for FVL, homozygous for PT20210, double heterozygous for FVL and PT20210, or deficient in PC, PS, or AT, existing data suggest a stronger association with arterial thrombotic risk. Given the in vivo role of these coagulation factors in the plasmatic coagulation pathway, we consider the use of anticoagulation (with or without aspirin) if these thrombophilias are identified in a patient with low bleeding risk.
Importantly, the patient’s thrombotic event was 1 month prior and he was not on anticoagulation at the time of evaluation, so testing functional assays (PC, PS, AT, LA) were expected to provide accurate results. FVL and PT mutation testing were sent to investigate for homozygous mutations; the patient was wild-type. Given his young age, PC, PS, and AT activities were tested and returned normal. LA and APLA (aCL and aβ2GPI IgG/IgM) also returned normal. CBC was repeated and in the absence of hematologic abnormalities, MPN and PNH were considered unlikely. However, given limited data on how often these disorders might cause an unexplained arterial event, JAK2V617F and flow cytometry for PNH were sent and returned normal.
In the absence of another contributing etiology, the patient’s event was classified as ESUS and continuation of antiplatelet therapy (aspirin 81 mg) for secondary prevention of ischemic stroke was recommended.
Case 2: 42-year-old woman with central retinal artery occlusion
A 42-year-old woman developed stuttering onset over a few hours and then almost complete painless visual loss in her left eye, prompting presentation to the emergency department. Fundoscopic examination revealed diffuse retinal pallor with associated arterial attenuation, consistent with central retinal artery occlusion. The patient was given aspirin and admitted to the inpatient stroke unit. Normal perfusion returned within 24 hours. She had no prior medical problems and denied family history of stroke or other thrombosis. Basic laboratory testing, urine drug screen, and screening for atherosclerotic risk factors (lipid panel, lipoprotein(a), hemoglobin A1c) were normal. ECG was without abnormality and TTE with bubble study was negative for PFO, intracardiac thrombus, or valvular disease. Bilateral duplex carotid ultrasounds were without luminal stenosis. MRI and magnetic resonance angiography of the brain and neck were unremarkable. The primary team ordered a thrombophilia workup including FVL, PT20210, PC, PS, and AT activities; all returned without abnormality. LA was strongly positive and APLA testing revealed aCL IgG 75 IgG phospholipid units (normal <23), IgM 19 IgM phospholipid units (normal <11), and aβ2GPI IgG >100 IgG phospholipid units (normal <20), IgM 11 IgM phospholipid units (normal <20). Hematology was consulted for diagnosis and management of possible APS.
Approach to case 2.
The patient has a documented central retinal artery occlusion. Workup to exclude atherosclerotic and cardioembolic sources was performed and revealed no abnormality. No causative medications or substances, signs of other systemic disease, or visible anatomic or vascular abnormalities were identified. Contralateral retinal examination by the ophthalmologist was normal. Therefore, the only risk factor identified was APLA, “triple positive.” To meet diagnostic criteria, APLA laboratory values must be repeated in 12 weeks,67 but the triple positivity and the strikingly high positive titers in the presence of an unexplained arterial thrombosis was highly suggestive of APS.
Optimal therapy for arterial thromboembolism in APS is unknown. Anticoagulation alone, antiplatelet therapy alone, or the combination can be used, but no consensus exists.94-96 We are aware of only 1 small randomized trial that compared treatment with aspirin 100 mg once daily alone to combination aspirin and vitamin K antagonist (target international normalized ratio, 2.0-3.0).97 Aspirin alone was associated with a higher incidence of stroke recurrence, with similar incidence of hemorrhagic complications. Two randomized trials comparing rivaroxaban with warfarin showed excess thrombotic events, mostly arterial, in the rivaroxaban arms.98,99 Therefore, if anticoagulation is considered in APS, DOACs are best avoided pending further investigation.
Consideration of bleeding risk is an essential part of the management decision in any patient, particularly when combination anticoagulation and antiplatelet therapy is considered. An incremental increase in bleeding risk, including major bleeding, when using combined antiplatelet and anticoagulant therapy has been shown in the cardiology and venous thromboembolism literature,100-102 although this increase has not been consistently found.97,103 Importantly, patients in these trials were older and had comorbid diseases, likely increasing their bleeding risk compared with the younger, otherwise healthy patient with unexplained arterial thrombosis.
A detailed discussion with the patient started with an acknowledgment of the lack of evidence to guide antithrombotic drug selection. We then considered the balance between her thrombotic and bleeding risk; she has markedly elevated APLA, is “triple positive,” and has no risk factors for bleeding. The patient shared our concern that her thrombosis risk, and the potential devastating consequences of a recurrent arterial event, were higher than her bleeding risk, so the joint decision was made to start anticoagulation with warfarin (international normalized ratio, 2.0-3.0) along with aspirin 81 mg daily, with planned reevaluation in 3 months.
Case 3: 25-year-old woman with renal infarct
A 25-year-old woman presented to the emergency department with sudden-onset left flank pain. Vitals signs were notable for blood pressure of 162/93 mm Hg. Serum creatinine was normal; urinalysis showed 2+ blood, 1+ protein, and many red blood cells. CT with contrast of the abdomen and pelvis revealed an infarct of the lower pole of the left kidney. She was started on a heparin drip. She denied any prior medical problems. She had been taking an oral contraceptive, drospirenone/ethinyl estradiol 3 mg/0.03 mg, since age 19. Initial workup included an ECG and cardiac monitor during hospitalization, and neither identified an arrhythmia. TTE was negative for intracardiac thrombus or valvular disease. Hematology was consulted during hospitalization because of concern for thrombophilia.
Approach to case 3.
The first step was to clarify whether the etiology of the renal infarct was arterial or venous occlusion; review of imaging with a radiologist indicated a wedge-shaped infarct, suggestive of an arterial event.
In a large case series, >50% of renal infarctions occurred because of cardiogenic embolism, but up to 30% of cases were idiopathic.104 Atherosclerotic risk assessment with lipid panel, lipoprotein(a), hemoglobin A1c, and smoking history, TTE with bubble study, and 30-day cardiac monitor were obtained and were unremarkable. Vessel wall abnormalities resulting from renal artery dissection or trauma104 or from uncommon disorders such as fibromuscular dysplasia105 and segmental arterial mediolysis106 can cause renal infarction. Careful review of the CT scan with an expert radiologist identified no vessel wall abnormalities. Because thromboembolism may arise from the aorta and better definition of the renal arteries was desirable, an aortic dissection protocol CT arteriogram was ordered, but was without abnormality. Laboratory data, clinical history, and physical examination provided no suspicion for a hematologic disorder, malignancy, or autoimmune disease.
Arterial thromboembolism associated with COC is uncommon but well described.17 Although data in venous thrombosis suggest that estrogen-containing therapies can be safely continued after thrombosis in patients who continue anticoagulation,107 a similar investigation has not been performed in arterial thrombosis. Given the lack of safety data and the availability of nonestrogen contraceptives, our preference is to switch to nonestrogen-based contraceptives after COC-associated arterial thrombosis.
No studies have assessed the role of antiplatelet therapy vs anticoagulation for patients with unexplained renal artery thrombosis and, therefore, clinical decisions are empiric and non-evidence based. Published case series report use of anticoagulation alone, aspirin alone, and anticoagulation for a limited time followed by long-term antiplatelet therapy.108,109 As discussed in case 2, an increased bleeding risk with combination therapy must be considered and weighed against recurrent thrombosis risk. Given the patient’s low risk for bleeding and the potential risk of thrombosis progression or recurrence, anticoagulation with warfarin plus aspirin was chosen, with close hematology follow-up.
At 3 months, we discussed the risks and benefits of anticoagulation cessation and the potential utility of thrombophilia testing. Thrombophilia tests not influenced by anticoagulation (FVL, P20201, aCL IgG/IgM, aβ2GPI IgG/IgM) were obtained; she was found to be heterozygous for FVL, which did not influence her management plan (Table 5). The risk of recurrent arterial thrombosis after a COC-associated event is unknown, but given the patient had stopped her COC and had no other identifiable risk factors, anticoagulation was suspended and aspirin continued. Four weeks later, the patient returned for the remainder of thrombophilia testing (PC, PS, AT, LA), which returned normal. She remained off anticoagulation; aspirin was continued long-term.
Conclusion
The workup and management of unexplained arterial thrombosis is complex because of the multiple potential contributors to arterial thrombotic risk and the lack of data on recommended diagnostic evaluation, thrombosis recurrence risk, and best management. Further research is needed to define the role of thrombophilias in arterial events and to identify which agents are superior for secondary prevention. Nevertheless, with a structured approach (Tables 2 and 3), hematologists can assist subspecialists in ensuring that a comprehensive evaluation is performed and can then facilitate an informed conversation with the patient to select the most appropriate treatment plan.
Authorship
Contribution: J.E.M. developed the concept and design of the manuscript, wrote the manuscript, and gave final approval; and S.M. developed the concept and design of the manuscript, wrote the manuscript, and gave final approval.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stephan Moll, University of North Carolina School of Medicine, Department of Medicine, Division of Hematology, CB 7035, Chapel Hill, NC 27599; e-mail: smoll@med.unc.edu; or Jori E. May, University of Alabama at Birmingham, Department of Medicine, Division of Hematology/Oncology, 1720 2nd Ave South, NP 2540, Birmingham, AL 35294; e-mail: jemay@uabmc.edu.
