

In this issue of Blood, show that 2 splicing factor mutations can coexist in the same cell in myeloid malignancy patients when their combination includes less common mutant alleles.1
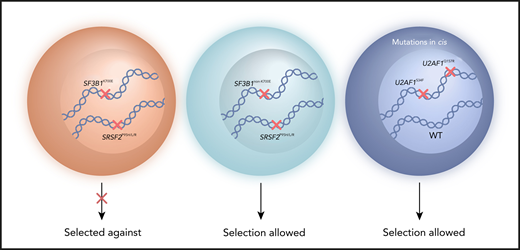
Myeloid malignancy patient cells with 2 common splicing factor mutations, such as SF3B1K700E and SRSF2P95H/L/R mutations, are selected against. Selection can occur instead for cells with 2 splicing factor mutations that include less common alleles, such as SF3B1non-K700E mutations (or rare amino acid changes at SRSF2 P95). Selection of cells with combined U2AF1S34 and U2AF1Q157 mutations has been shown when the 2 mutations occur in cis with preservation of 1 wild-type U2AF1 allele. WT, wild-type.
Myeloid malignancy patient cells with 2 common splicing factor mutations, such as SF3B1K700E and SRSF2P95H/L/R mutations, are selected against. Selection can occur instead for cells with 2 splicing factor mutations that include less common alleles, such as SF3B1non-K700E mutations (or rare amino acid changes at SRSF2 P95). Selection of cells with combined U2AF1S34 and U2AF1Q157 mutations has been shown when the 2 mutations occur in cis with preservation of 1 wild-type U2AF1 allele. WT, wild-type.
Mutations in genes involved in pre-mRNA splicing (SF3B1, SRSF2, and U2AF1 mutations being the most frequent) are common in patients with myeloid malignancies, including myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), and myeloproliferative neoplasms.2,3 Several studies involving the analysis of large patient cohorts demonstrated that splicing factor mutations are typically heterozygous, and a striking and consistent finding of these studies is that these mutations are mutually exclusive.4-6 Nevertheless, myeloid malignancy patients harboring 2 cooccurring splicing factor mutations have been reported, albeit rarely. The existence of such cases represents a conundrum, since the combination of 2 splicing factor mutations would be expected to be lethal for a cell, unless each mutation occurs in different clones. Indeed, a previous study demonstrated that coexpression of the most common mutations of SF3B1 (K700E) and SRSF2 (P95H) in vivo in mice is intolerable to hematopoietic cells.7
Until now, the characteristics of patients with myeloid malignancies harboring multiple splicing factor mutations had not been systematically investigated. In their study, Taylor et al performed bulk DNA sequencing and single-cell sequencing of malignant cell populations from myeloid malignancy patients to determine the frequency and basis for the co-existence of splicing factor mutations.
The authors analyzed genomic DNA sequencing data from more than 4,000 myeloid malignancy patients, and mutations of SF3B1, SRSF2, U2AF1 and ZRSR2 were observed in approximately 23% of all cases. They identified 36 cases with 2 cooccurring splicing factor mutations. After inclusion of 22 additional patients (from the Mayo Clinic) with 2 splicing factor mutations, the analysis of the variant allele frequency and cancer cell fraction showed that the mutations coexisted within the same cell in approximately two thirds of the overall cohort of 58 double-mutant samples. Intriguingly, SF3B1K700 and SRSF2P95/P96 mutations, which represent the most frequent mutant alleles among splicing factors in myeloid malignancies, were significantly less common in double mutants compared with single mutants, indicating selection against cells with cooccurrence of SF3B1K700 and SRSF2P95/P96 mutations. In contrast, selection was observed for less common alleles, such as SF3B1non-K700E mutations (eg, E622, H662, K666), rare amino acid substitutions at SRSF2 P95, and combined U2AF1S34/Q157 mutations (see figure). These data support the conclusion of the study that mutual exclusivity or cooccurrence of splicing factor mutations is allele specific rather than gene specific.
Taylor et al proceeded to perform single-cell DNA sequencing of bone marrow cells from 11 patients that harbored 2 splicing factor mutations. The data elegantly confirmed the mutual exclusivity of SF3B1K700E and SRSF2P95H mutations at the single-cell level and demonstrated the potential for cooccurrence of other rare splicing factor–mutant alleles.
Functional studies involving the measurement of the extent of missplicing events for SF3B1 mutants and the binding of SRSF2 mutants to its consensus RNA sequence were then carried out. The proteins encoded by the less common SF3B1 and SRSF2 mutant alleles that are enriched in double-mutant myeloid malignancy patients were shown to have reduced effects on RNA splicing or binding affinity compared with the most common alleles, providing evidence supporting the concept that the less common mutant alleles may escape from epistasis due to more modest effects on RNA binding and/or splicing.
Interestingly, it was shown that U2AF1S34 and U2AF1Q157 mutations cooccurred in myeloid malignancy patients at a significantly higher frequency than expected by chance. Single-cell DNA sequencing analysis of a double-mutant patient showed that both U2AF1S34 and U2AF1Q157 mutations were present in the same cells, suggesting potential cooperation between the 2 mutations. Furthermore, these U2AF1 mutations were found to cooccur in cis with preservation of the wild-type allele, a finding in agreement with a previous study demonstrating that the expression of the wild-type U2AF1 allele is required for survival of cells harboring a U2AF1 mutation.8 The analysis of further double-U2AF1-mutant patient samples is required to establish whether U2AF1S34 and U2AF1Q157 mutations are tolerable when cooccurring in trans.
This study by Taylor et al has illuminated the genetic and molecular bases for the escape of splicing factor mutations from epistasis in patients with myeloid malignancies, findings that have important clinical and therapeutic implications.
Specific mutant alleles of each splicing factor gene might have different impacts on the clinical features and/or survival of patients with myeloid malignancies. Indeed, this suggestion is supported, for example, by a recent study showing that SF3B1K666 mutations are associated with some hematological features in MDS and with shorter patient survival and increased progression to AML.9 However, the observation by Taylor et al that SF3B1K666 mutations have a weaker effect on pre-mRNA splicing than SF3B1K700 mutations, likely resulting from distinct structural disturbances at these amino acid locations, might be expected to lead to a milder impact of SF3B1K666 mutations on patient outcome. Further studies, including functional assessment of aberrantly spliced target genes, are required to elucidate fully the effects of less common splicing factor–mutant alleles on clinical features and outcome in patients with myeloid malignancies.
The mutual exclusivity of splicing factor mutations, previous studies showing that these mutations are not tolerated in a homozygous state,7 and the demonstration that the survival of splicing factor–mutant cells depends on presence of the wild-type allele8 provided the rationale for the potential therapeutic use of splicing modulators in splicing factor–mutant myeloid malignancy patients. The basis of this synthetic lethality strategy is that, unlike wild-type cells, splicing factor–mutant cells would be unable to tolerate further disruption to the splicing process by pharmacological inhibition of the spliceosome.10 The finding by Taylor et al that the most common SF3B1 and SRSF2 mutations have more prominent effects on pre-mRNA splicing and RNA-binding affinity than less common splicing factor–mutant alleles indicates that myeloid malignancy patients with SF3B1K700E or SRSF2P95H/L/R mutations may be more susceptible to treatment with splicing modulators. Stratification of patients on the basis of specific splicing factor–mutant alleles should be considered in clinical trials involving drugs that target the spliceosome.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal