

TO THE EDITOR:
The thrombotic spectrum of antiphospholipid syndrome (APS) is heterogeneous and ranges from mild thrombosis in isolation to a catastrophic, multisystem disease, with thrombotic microangiopathy and organ failure. The exact mechanism of APS-related thrombosis remains to be elucidated and may differ between subsets of patients. Murine data have linked thrombosis, at least in part, to complement activation.1,2 Chaturvedi et al,3 in their interesting article in Blood, reported that patients’ serum induced C5b9 formation on human endothelial hybrid cells and corresponding complement-dependent cell killing (ie, modified Ham test). The authors argued that, with increasing severity of disease, the role of unrestrained complement activation via the alternative pathway becomes more dominant, as corroborated by the high prevalence of complement gene variants in catastrophic APS, quite like primary atypical hemolytic uremic syndrome (aHUS). Their data suggest that such patients should be screened for genetic variants and treated accordingly.4
We evaluated the premise of Chaturvedi et al in an intrinsically different subset of patients with APS,5 who presented with APS nephropathy in a kidney biopsy.6 Serum samples were obtained, processed, and immediately stored at −80°C at the time of the biopsy. We used human microvascular endothelial cells (ATCC, Manassas, VA).7 C5b9 formation on these endothelial cells reflects the dynamics of complement and disease activity in patients with primary aHUS.8,9 In brief, perturbed endothelial cells were plated on glass culture slides, incubated with serum diluted in medium, and stained for C5b9; pooled normal human serum and serum from patients with primary aHUS were run in parallel.8 In selected experiments, the endothelium was stained for C3c or immunoglobulin G (IgG) subclasses. Furthermore, we analyzed in vivo complement activation. Kidney sections were stained for C1q, C4d, C3c, and C5b9 to dissect the complement cascade. Methods are detailed in the supplemental Material and methods (available on the Blood Web site). The study was approved by the regional ethics committee and was performed in accordance with the Declaration of Helsinki.
In total, 17 consecutive patients with APS nephropathy were included (Table 1). The female-to-male ratio was 0.9, and the median age at diagnosis was 45 (interquartile range, 27-55) years. Patients invariably presented with proteinuric kidney disease (nephrotic-range proteinuria, n = 4), either with (n = 6) or without microscopic hematuria. Kidney tissue sections showed both acute and chronic morphologic features of thrombotic microangiopathy in 14 cases; subendothelial electron-lucent material confirmed the thrombotic microangiopathy on electron microscopy (n/N = 7/8). Patients 01, 02, and 04 had focal cortical necrosis without glomerular lesions, reflecting arteriolar thrombosis. Four (24%) patients were triple positive, 8 (47%) were double positive, and 5 (29%) were single positive for lupus anticoagulant, anti-β2 glycoprotein-1 antibodies, or anti-cardiolipin antibodies, confirmed on 2 occasions at least 12 weeks apart. Of note, thrombocytopenia, but not hemolytic anemia, was found in 8 (47%) patients. C4 and C3 levels were low in 2 (n = 14; 14%) and 4 (n = 14; 29%) patients, respectively. The functional activity of the classic pathway was decreased in 6 of 13 (46%) patients. None of the patients had systemic lupus erythematosus.
Baseline characteristics of 17 patients with APS and APS nephropathy on kidney biopsy
| APS diagnostic criteria* | Ex vivo C5b9 formation | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Kidney biopsy, y | Laboratory | Extrarenal manifestations (y) | Age, y | Sex | SCr, µmol/L | uRBC | uP, g/d | Plts, g/L | C4, g/L (0.11)† | C3, g/L (0.75)† | CPFA, % (75)† | % vs normal | P |
| 01 | 1984 | aCL | — | 48 | M | 80 | – | 1.90 | WNR | 0.30 | 1.36 | >125 | Normal | NS |
| 02 | 1985 | aCL | — | 45 | M | 217 | – | 1.94 | WNR | 0.26 | 1.10 | >125 | 60 | NS |
| 03 | 1985 | LAC, β2GPI, aCL | CAPS‡ (1986) | 50 | M | 223 | – | 0.78 | 93 | ND | ND | ND | ND | |
| 04 | 1986 | β2GPI, aCL | AT (2001), pregnancy | 28 | F | 75 | – | 1.11 | WNR | 0.14 | 0.78 | ND | 109 | NS |
| 05 | 1988 | aCL | AT (2004) | 38 | M | 214 | – | 1.02 | 314 | 0.20 | 1.30 | 3 | 63 | NS |
| 06 | 1988 | LAC, aCL | AT (1990) | 58 | F | 168 | + | 0.38 | 108 | 0.07 | 0.55 | 58 | 117 | NS |
| 07 | 1999 | β2GPI, aCL | VT (1995) | 26 | F | 657 | + | 1.85 | 83 | ND | ND | ND | ND | |
| 08 | 1999 | LAC, β2GPI, aCL | VT (1995) | 53 | M | 155 | – | 4.80 | 93 | 0.18 | 1.04 | 47 | 127 | NS |
| 09 | 2000 | LAC, β2GPI, aCL | — | 26 | M | 119 | – | 0.06 | 152 | 0.15 | 0.93 | ND | 161 | NS |
| 10 | 2000 | LAC, aCL | Pregnancy | 45 | F | 197 | – | 1.20 | 101 | 0.09 | 0.60 | 61 | 189 | .01 |
| 11 | 2002 | aCL | — | 62 | M | 216 | + | 3.64 | 333 | 0.11 | 0.72 | 57 | 103 | NS |
| 12 | 2005 | β2GPI, aCL | — | 73 | F | 284 | + | 1.02 | 97 | 0.13 | 0.73 | 103 | 101 | NS |
| 13 | 2007 | aCL | — | 67 | M | 276 | + | 13.06 | WNR | 0.37 | 1.18 | 122 | 96 | NS |
| 14 | 2007 | β2GPI, aCL | VT (2002) | 27 | F | 69 | + | 0.48 | 314 | 0.16 | 1.09 | 71 | 71 | NS |
| 15 | 2010 | LAC, aCL | — | 19 | F | 103 | – | 0.21 | WNR | 0.11 | 1.02 | 109 | 160 | NS |
| 16 | 2012 | β2GPI, aCL | — | 47 | M | 188 | – | 3.95 | 101 | 0.12 | 1.03 | 33 | 123 | NS |
| 17 | 2014 | LAC, β2GPI, aCL | VT (1998), AT‡ (2014) | 39 | F | 266 | – | 2.72 | 51 | ND | ND | >125 | 78 | NS |
| APS diagnostic criteria* | Ex vivo C5b9 formation | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Kidney biopsy, y | Laboratory | Extrarenal manifestations (y) | Age, y | Sex | SCr, µmol/L | uRBC | uP, g/d | Plts, g/L | C4, g/L (0.11)† | C3, g/L (0.75)† | CPFA, % (75)† | % vs normal | P |
| 01 | 1984 | aCL | — | 48 | M | 80 | – | 1.90 | WNR | 0.30 | 1.36 | >125 | Normal | NS |
| 02 | 1985 | aCL | — | 45 | M | 217 | – | 1.94 | WNR | 0.26 | 1.10 | >125 | 60 | NS |
| 03 | 1985 | LAC, β2GPI, aCL | CAPS‡ (1986) | 50 | M | 223 | – | 0.78 | 93 | ND | ND | ND | ND | |
| 04 | 1986 | β2GPI, aCL | AT (2001), pregnancy | 28 | F | 75 | – | 1.11 | WNR | 0.14 | 0.78 | ND | 109 | NS |
| 05 | 1988 | aCL | AT (2004) | 38 | M | 214 | – | 1.02 | 314 | 0.20 | 1.30 | 3 | 63 | NS |
| 06 | 1988 | LAC, aCL | AT (1990) | 58 | F | 168 | + | 0.38 | 108 | 0.07 | 0.55 | 58 | 117 | NS |
| 07 | 1999 | β2GPI, aCL | VT (1995) | 26 | F | 657 | + | 1.85 | 83 | ND | ND | ND | ND | |
| 08 | 1999 | LAC, β2GPI, aCL | VT (1995) | 53 | M | 155 | – | 4.80 | 93 | 0.18 | 1.04 | 47 | 127 | NS |
| 09 | 2000 | LAC, β2GPI, aCL | — | 26 | M | 119 | – | 0.06 | 152 | 0.15 | 0.93 | ND | 161 | NS |
| 10 | 2000 | LAC, aCL | Pregnancy | 45 | F | 197 | – | 1.20 | 101 | 0.09 | 0.60 | 61 | 189 | .01 |
| 11 | 2002 | aCL | — | 62 | M | 216 | + | 3.64 | 333 | 0.11 | 0.72 | 57 | 103 | NS |
| 12 | 2005 | β2GPI, aCL | — | 73 | F | 284 | + | 1.02 | 97 | 0.13 | 0.73 | 103 | 101 | NS |
| 13 | 2007 | aCL | — | 67 | M | 276 | + | 13.06 | WNR | 0.37 | 1.18 | 122 | 96 | NS |
| 14 | 2007 | β2GPI, aCL | VT (2002) | 27 | F | 69 | + | 0.48 | 314 | 0.16 | 1.09 | 71 | 71 | NS |
| 15 | 2010 | LAC, aCL | — | 19 | F | 103 | – | 0.21 | WNR | 0.11 | 1.02 | 109 | 160 | NS |
| 16 | 2012 | β2GPI, aCL | — | 47 | M | 188 | – | 3.95 | 101 | 0.12 | 1.03 | 33 | 123 | NS |
| 17 | 2014 | LAC, β2GPI, aCL | VT (1998), AT‡ (2014) | 39 | F | 266 | – | 2.72 | 51 | ND | ND | >125 | 78 | NS |
aCL, anti-cardiolipin antibodies; AT, arterial thrombosis; β2GP1, anti-beta2 glycoprotein-1 antibodies; CAPS, catastrophic APS; CPFA, classical pathway functional activity; F, female; LAC, lupus anticoagulant; M, male; ND, not determined; NS, not significant; SCr, serum creatinine; uP, proteinuria; uRBC, hematuria; VT, venous thrombosis; WNR, within normal range.
Diagnostic criteria are from Miyakis et al.5
Lower limit of normal.
12 mo from kidney biopsy.
At the time of presentation, 14 (93%) of 15 patients with APS nephropathy showed normal ex vivo C5b9 formation on the perturbed endothelium (Figure 1A), whereas massive ex vivo C5b9 formation was found in 5 patients with aHUS and a pathogenic gain-of-function variant in C3 (ie, c.481C>T; p.R161W)10 was run in parallel. To date, none of our patients with primary aHUS and at least 1 genetic variant linked to complement dysregulation of the alternative pathway had normal ex vivo test results.11 Ex vivo C3c deposits appeared normal in the setting of APS nephropathy in 4 samples examined, excluding complement activation upstream of C5.
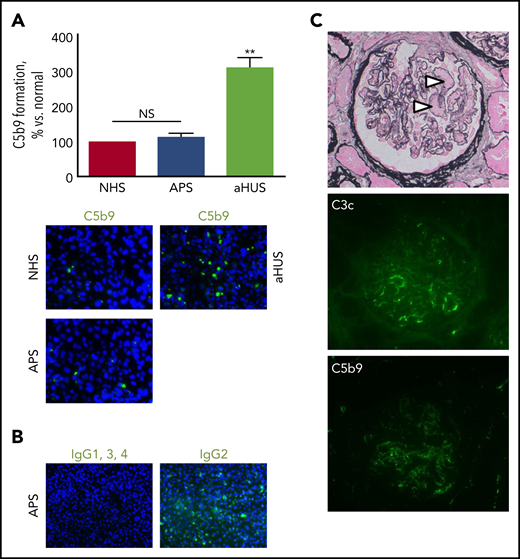
Patients with APS and microvascular thrombosis identified by kidney biopsy have normal ex vivo complement activation and lack significant in vivo complement deposits. (A) Ex vivo C5b9 formation on the perturbed endothelium did not differ between patients with APS (n = 15) and normal human serum (NHS), whereas serum from patients with primary aHUS and a pathogenic gain-of-function variant in C3 induced unrestrained complement activation (n = 5); original magnification ×400. **P < .01; NS not significant. (B) IgG2, but not the other subclasses, bound to the endothelium when incubated with serum from triple-positive patients (2 of 2); original magnification ×200. (C) Microvascular thrombosis on kidney biopsy (arrowheads). Scant deposits of C3c (7 of 17) and/or C5b9 (2 of 17) along segments of the glomerular capillary wall were uncommon. Original magnification ×400.
Patients with APS and microvascular thrombosis identified by kidney biopsy have normal ex vivo complement activation and lack significant in vivo complement deposits. (A) Ex vivo C5b9 formation on the perturbed endothelium did not differ between patients with APS (n = 15) and normal human serum (NHS), whereas serum from patients with primary aHUS and a pathogenic gain-of-function variant in C3 induced unrestrained complement activation (n = 5); original magnification ×400. **P < .01; NS not significant. (B) IgG2, but not the other subclasses, bound to the endothelium when incubated with serum from triple-positive patients (2 of 2); original magnification ×200. (C) Microvascular thrombosis on kidney biopsy (arrowheads). Scant deposits of C3c (7 of 17) and/or C5b9 (2 of 17) along segments of the glomerular capillary wall were uncommon. Original magnification ×400.
We consider the kidney tissue sections from our patients, with the presence of abundant endothelial cells, to be an ideal in vivo counterpart for the study of complement activation, either with coinciding immunoglobulin deposition or not, in relation to local thrombotic vascular changes. Seven (41%) of 17 samples revealed scant C3c deposits along segments of the glomerular capillary wall; C3c colocated with C5b9 in 2 cases (Figure 1C). Neither C1q nor C4d was found, making activation of the classic and lectin pathways highly unlikely. No electron-dense deposits were found on electron microscopy, underscoring the lack of complement deposits.
The clinical course and kidney survival of our patients also differed from the dire prognosis of primary aHUS not treated with therapeutic complement inhibition.12,13 Patients were followed up for a median of 5.4 (interquartile range, 1.0-16.8) years; 2 patients were lost to follow-up. The follow-up is depicted in supplemental Table 1. At presentation, anticoagulation and immunosuppression were started in 10 (67%) and 7 (47%) of 15 patients, respectively. None of the patients received therapeutic complement inhibition. Kidney function stabilized and/or improved in 13 (87%) of 15 patients. Patients 07 and 02, who presented with end-stage kidney disease, did not recover kidney function. Five patients had recurrent thrombosis, including the 4 patients who did not receive anticoagulation; 1 of the latter (patient 03) died of catastrophic APS.
Together, our experimental and clinical findings suggest that a mechanism other than unrestrained complement activation is key for the occurrence of renal thrombosis in APS. We studied the dynamics of complement activation on the endothelium during active arteriolar and/or microvascular thrombosis, whereas the time of sampling in the cohort in Chaturvedi et al3 did not concur with the (macrovascular) thrombotic event. In addition, the modified Ham test uses endothelial hybrid cells that lack the complement regulatory proteins CD55 and CD59, with a lower threshold for complement activation, as compared with our ex vivo test. Based on our observations, we decided not to test for variants in complement genes.7,11 Also, it remains to be established whether the reported variants in complement genes3 are causal of (macrovascular) thrombosis. First, the minor allele frequency of some variants exceeds 0.1%; for example, deletion of CFHR1 and CFHR3 has been identified in up to 8% of the European population,14 indicating a nonpathogenic change in the absence of factor H autoantibodies. Second, loss of function variants in CFB and CFHR5 are of no significance, as the transcribed proteins are unlikely to overactivate complement.15 Third, the effects of variants in CFHR4, THBD, and DGKE on complement regulation are still controversial.
What then is the mechanism of APS nephropathy? Experimental data in mice showed that non–complement-dependent activation of tissue factor is key for the occurrence of renal thrombosis.2 This fits our observation that non–complement-fixing IgG2 was found on the endothelium after serum incubation, whereas complement-fixing IgG1 and IgG3 were not found in either of 2 patients (Figure 1B). APS-related thrombosis, indeed, has been linked to anti-β2 glycoprotein-1 and anti-cardiolipin IgG2, but not to other IgG subclasses.16,17 We therefore assume that the antiphospholipid antibodies may cause renal thrombosis via a direct effect on the endothelium.18 In addition, annexin A5 resistance may play a role in a subset of patients, including those with anti-β2 glycoprotein-1 antibodies.19
In summary, the suggestion that unrestrained complement activation on the endothelium correlates with thrombosis in APS cannot be extrapolated to patients with APS nephropathy. Future studies are needed on the role of complement in various subsets of patients with APS.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors gratefully thank N. Bijnens, P. van Breda-Vriesman, H. van Rie, and R. Theunissen for excellent technical assistance (Maastricht University Medical Center, Maastricht, The Netherlands).
Authorship
Contribution: S.A.M.E.G.T., C.P.R., and P.v.P. designed the research and analyzed the data; and S.A.M.E.G.T. wrote the primary manuscript with assistance and editing by J.G.M.C.D., C.P.R., and P.v.P.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the Limburg Renal Registry appears in “Appendix.”
Correspondence: Pieter van Paassen, Department of Nephrology and Clinical Immunology, Maastricht University Medical Center, P. Debyelaan 25, 6229 HX Maastricht, The Netherlands; e-mail: p.vanpaassen@maastrichtuniversity.nl.
Appendix
The members of the Limburg Renal Registry are: F. de Heer, M. Krekels, F. Stifft, G. Verseput (Zuyderland Medical Center, Sittard-Geleen, The Netherlands); S. Boorsma, W. Grave, J. Huitema, J. Wirtz (St. Laurentius Hospital, Roermond, The Netherlands); N. ter Braak, L. Frenken, and S. Gaertner (Zuyderland Medical Center, Heerlen, The Netherlands); and M. Christiaans, T. Fung, M. Gelens, J. Kooman, K. Leunissen, E. Litjens, J. van der Net, F. van der Sande, and E. van Duijnhoven (Maastricht University Medical Center, Maastricht, The Netherlands).

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal