


Abstract
Activated phosphatidylinositol 3-kinase-δ (PI3K-δ) syndrome (APDS) is a rare primary combined immunodeficiency caused by either dominant gain-of-function mutations in the PIK3CD gene encoding the catalytic subunit p110δ of PI3K-δ (referred to as type 1 APDS) or dominant loss-of-function mutations in the PIK3R1 gene encoding the p85α, p55α, and p50α regulatory subunits (type 2 APDS). In types 1 and 2 APDS, the PI3K-δ hyperactivity resulting from the gene mutations leads to similar clinical presentations, characterized by increased susceptibility to bacterial and viral infections and (to a lesser extent) autoimmune manifestations. A hallmark of this disease is lymphoproliferation, which may even be life threatening and require repeated surgical treatment. A major complication of APDS is malignancy (especially B-cell lymphomas), which greatly worsens the prognosis. Here, we review the different neoplastic conditions observed in patients with APDS and discuss the uncontrolled PI3K-δ activity in B and T cells that leads to malignant transformation.
Introduction
Recent experiments in mammals have shown that balanced class 1A phosphatidylinositol 3-kinase (PI3K) activity in lymphocytes is required for an optimal immune response. The PI3K protein complex comprises a p110 catalytic subunit (p110α, p110β, or p110δ) and a regulatory subunit (p85α, p55α, p50α, p85β, or p55γ). The p110δ subunit is mainly expressed in leukocytes, where it predominantly interacts with the ubiquitously expressed regulatory subunit p85α. The class 1A PI3Ks convert phosphatidylinositol 4,5-bisphosphate into phosphatidylinositol 3,4,5-trisphosphate, an important phospholipid secondary messenger. Each of the catalytic subunits can bind to any of the regulatory subunits and thus respond to extracellular signals (B- or T- cell receptors, cytokine receptors, and costimulatory molecules) by influencing cell cycle progression, metabolic control, and cell growth, survival, and migration.1 The regulatory subunit is required for proper activity of the catalytic subunit, regulating its stability, cellular localization, and function. Hence, defective PI3K-δ signaling caused by autosomal recessive loss-of-function mutations in PIK3CD (coding for p110δ) results in a combined immunodeficiency characterized by low B-cell, natural killer (NK)–cell, and T-cell counts and impaired lymphocyte function.2 Biallelic mutations in PIK3CD have been recently reported in several patients.3-6 Homozygous nonsense mutations in PIK3R1 (affecting only the p85α subunit) impair B-cell development and function and result in an autosomal recessive agammaglobulinemic phenotype.7,8
In contrast, increased PI3K-δ signaling leads to a lymphoproliferation-associated combined immunodeficiency with autosomal dominant transmission. Two main disorders have been described to date. Type 1 activated PI3K syndrome (APDS1; Online Mendelian Inheritance in Man [OMIM] # 61553; also termed immunodeficiency 14 [IMD14] or p110-δ-activating mutation causing senescent T cells, lymphadenopathy, and immunodeficiency [PASLI]) is due to heterozygous gain-of-function (GOF) mutations in PIK3CD that can affect different domains in the encoded protein. The most frequent disease-causing mutation is located in exon 24 (coding for part of the kinase domain of PI3K) and leads to an E1021K amino acid substitution.9,10 Type 2 activated PI3K syndrome (APDS2; OMIM #616005; also termed IMD36 or PASLI-R1) is caused by heterozygous mutations affecting the splice sites of PIK3R1 exon 11.11,12 As a consequence of these splice site mutations, in-frame skipping of exon 11 (coding exon 10) results in a p85α protein lacking part of the iSH2 domain (Δ434-475 amino acids). In turn, a structural change in the shortened p85α disrupts the inhibitory interfaces in the nSH2, iSH2, and cSH2 domains that interact with p110δ.13 The p50α and p55α subunits are similarly affected.12
Both APDS1 and APDS2 lead to elevated activation of PI3K-δ, as shown by increased phosphorylation of the protein kinase B (AKT) located downstream in the PI3K signaling pathway (Figure 1). The clinical consequences of and biologic abnormalities in APDS1 and APDS2 are similar, although subtle differences can be found.
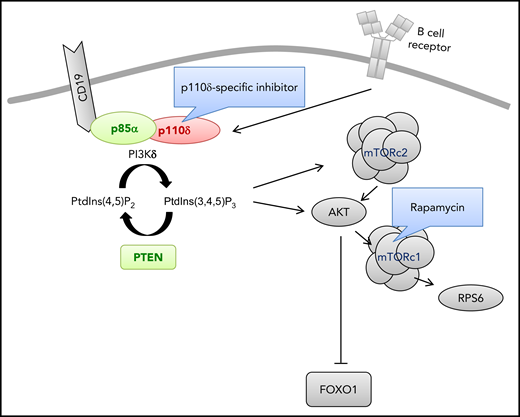
Genetic aberrations causing primary immunodeficiencies and hyperactivated PI3K-δ signaling in B lymphocytes. The indicated gene products cause primary immunodeficiencies and hyperactivated PI3K-δ signaling in B lymphocytes as a result of GOF mutations (red) or loss-of-function mutations (green). Drug treatments intended to diminish activated PI3K-δ signaling are also shown. FOXO1, forkhead box protein O1; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mTOR complex 2; PtdIns(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PtdIns(3,4,5)P3, phosphatidylinositol (3,4,5)-trisphosphate; PTEN, phosphatase and tensin homolog; RPS6, ribosomal protein S6.
Genetic aberrations causing primary immunodeficiencies and hyperactivated PI3K-δ signaling in B lymphocytes. The indicated gene products cause primary immunodeficiencies and hyperactivated PI3K-δ signaling in B lymphocytes as a result of GOF mutations (red) or loss-of-function mutations (green). Drug treatments intended to diminish activated PI3K-δ signaling are also shown. FOXO1, forkhead box protein O1; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mTOR complex 2; PtdIns(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PtdIns(3,4,5)P3, phosphatidylinositol (3,4,5)-trisphosphate; PTEN, phosphatase and tensin homolog; RPS6, ribosomal protein S6.
The clinical features and severity of APDS1 and APDS2 vary greatly from 1 patient to another, even within the same family.14,15 Patients present with recurrent bacterial and viral infections, evidencing a combined T and B immunodeficiency. The respiratory tract is primarily affected, and bronchiectasis is common. The presence of combined immunodeficiency can be confirmed by immunologic profiling, with a decrease in the counts of naïve CD4 and naïve CD8 T cells and naïve, memory, and switched B cells. Increased counts of transitional B cells, terminally differentiated/senescent CD8+CD57+ T cells, and CD8 effector/memory T cells are additional hallmarks of APDS. Immunoglobulin profiles are heterogeneous and range from hyperimmunoglobulin M syndrome to panhypogammaglobulinemia with defective antibody production. In addition to immunodeficiency, patients also present with lymphoproliferation affecting the lymph nodes, adenoids, tonsils, and spleen. This is another hallmark of the disease and may require repeated surgical treatment. Lymph node biopsies from patients with APDS1 or APDS2 show hyperplasia of programmed cell death 1 (PD-1)–positive T follicular helper (TFH) cells in germinal centers (GCs) and extrafollicular areas. The GCs are ill defined and appear to be disrupted by infiltration of the PD-1+ T cells.14,15
Another reported complication is autoimmunity, which mostly affects blood cells and can lead to thrombocytopenic purpura and hemolytic anemia. Although neurodevelopmental delay has been noted in both APDS1 and APDS2, growth retardation is especially observed in patients with APDS215 and is reminiscent of short stature, hyperextensibility of joints, ocular depression, Rieger anomaly, and teething delay (SHORT) syndrome, a disorder caused by heterozygous missense mutations located downstream of exon 11 in the PIK3R1 gene.16-18 In contrast to APDS2, SHORT syndrome is associated with abnormally low PI3K signaling.16-18
Lastly, the most serious complication of APDS is neoplasia (especially B-cell lymphoma), which greatly worsens a patient’s prognosis.
Occurrence of neoplasia in APDS patients
In the 2 main reviews of the clinical features of patients with APDS, the incidence of malignant disease was 13% for APDS1 and 28% for APDS2.14,15 Recently, a retrospective evaluation of 56 APDS1 patients with confirmed germ line GOF mutations in PIK3CD (part of the National Institute of Allergy and Infectious Diseases cohort) found an incidence of 30%.19 Furthermore, B-cell lymphoma has been reported in several patients not included in these cohort studies, thus further highlighting the risk of B-cell lymphoma in patients with APDS1 or APDS2 (Table 1). The most frequent cancers are B-cell lymphomas (mainly classical Hodgkin lymphoma [CHL] or diffuse large B-cell lymphoma [DLBCL]). However, marginal zone B-cell lymphoma and mucosal-associated lymphoid tissue–derived lymphoma have also been reported. An association with Epstein-Barr virus (EBV) was reported in 5 of the 14 cases of CHL and 8 of the 12 cases of DLBCL. However, these numbers might be underestimates, because EBV status was not documented in several patients with B-cell lymphoma (Table 1). An EBV-induced polymorphic lymphoproliferative disorder reminiscent of posttransplantation lymphoproliferative syndrome has been observed in a small number of patients.19 Some patients developed 2 successive B-cell lymphomas, for example, early-onset CHL followed some years later by DLBCL. Single cases of lymphoplasmacytic lymphoma and chronic lymphocytic leukemia (CLL) have also been reported. Apart from B-cell malignancies, individual cases of primary cutaneous anaplastic large-cell lymphoma, enteropathy mastocytoma, and acute myeloid leukemia (possibly related to treatment of CHL) have been also described from time to time. B-cell lymphoma thus seems to be a frequent complication of APDS, and the cumulative risk of developing cancer at the age of 40 years was to be calculated 78% for those with APDS2.15 The mortality rate testifies to the severity of the cancer, because 16% of APDS patients die as a result of lymphoma-associated complications (Table 1).
Characteristics of B-cell lymphomas in patients with APDS
| Type of B-cell lymphoma | EBV status (n) | Total (APDS1/APDS2) n of patients | M/F sex | Median (range) age at lymphoma diagnosis, y* | Lymphoma-associated death | Reference |
|---|---|---|---|---|---|---|
| Marginal zone lymphoma | NR | 5 (4/1) | 0/5 | 15 (13-18) | NR | 9,15,35,63 |
| Mucosa-associated lymphoid tissue lymphoma | EBV− (3) | 5 (4/1) | 4/1 | 21 (11-32) | NR | 15,19-21 |
| NR (2) | ||||||
| Hodgkin lymphoma | EBV+ (5) | 14 (9/5) | 4/8† | 15 (2.5-27) | 2 | 10,12,14,15,19,20,54,62,64 |
| NR (9) | ||||||
| Diffuse large-cell lymphoma | EBV+ (8) | 18 (13/5) | 3/14† | 22 (6-63) | 5 | 10,14,15,19,20,35,52,54,63,65 |
| EBV− (4) | ||||||
| NR (6) | ||||||
| Non-Hodgkin lymphoma (not otherwise specified) | EBV+ (2) | 5 (4/1) | 3/2 | 16 (8-20) | 1 | 19,20,66 |
| EBV− (1) | ||||||
| NR (2) | ||||||
| Posttransplantation lymphoproliferative disorder-like disease | EBV+ (3) | 3 (3/0) | 1/2 | 21 (8-59) | NR | 19 |
| Type of B-cell lymphoma | EBV status (n) | Total (APDS1/APDS2) n of patients | M/F sex | Median (range) age at lymphoma diagnosis, y* | Lymphoma-associated death | Reference |
|---|---|---|---|---|---|---|
| Marginal zone lymphoma | NR | 5 (4/1) | 0/5 | 15 (13-18) | NR | 9,15,35,63 |
| Mucosa-associated lymphoid tissue lymphoma | EBV− (3) | 5 (4/1) | 4/1 | 21 (11-32) | NR | 15,19-21 |
| NR (2) | ||||||
| Hodgkin lymphoma | EBV+ (5) | 14 (9/5) | 4/8† | 15 (2.5-27) | 2 | 10,12,14,15,19,20,54,62,64 |
| NR (9) | ||||||
| Diffuse large-cell lymphoma | EBV+ (8) | 18 (13/5) | 3/14† | 22 (6-63) | 5 | 10,14,15,19,20,35,52,54,63,65 |
| EBV− (4) | ||||||
| NR (6) | ||||||
| Non-Hodgkin lymphoma (not otherwise specified) | EBV+ (2) | 5 (4/1) | 3/2 | 16 (8-20) | 1 | 19,20,66 |
| EBV− (1) | ||||||
| NR (2) | ||||||
| Posttransplantation lymphoproliferative disorder-like disease | EBV+ (3) | 3 (3/0) | 1/2 | 21 (8-59) | NR | 19 |
NR, not reported.
For cases in which age at lymphoma diagnosis was not reported, age at time of clinical report was taken into account.
Sex was not recorded in 1 or 2 cases.
Isolated case reports of nonleukocytic malignancies include basal cell carcinoma on the forehead,20 dysgerminoma,21 rhabdomyosarcoma,22 and papillary thyroid carcinoma23 for APDS1 patients and a papillary neoplasm in both breasts in an APDS2 patient.15 However, it should be noted that a vast majority of patients with APDS are diagnosed and observed in childhood, and so other neoplasms might occur in later life.
Physiopathogenesis of cancers in APDS
The predisposition to B-cell lymphoma in APDS might be caused by 1 or several abnormalities. Firstly, it has been clearly demonstrated that the catalytic and regulatory subunits of PI3K-δ are strongly expressed in B cells and have a key role in B-cell maturation. One can reasonably hypothesize that uncontrolled activation of the PI3K pathway in B cells (via an antigen-driven [B-cell receptor] or EBV trigger) could directly increase cell survival and malignant transformation. Increased activation of the conserved serine/threonine kinase mTOR (a downstream effector of the PI3K/AKT pathway) is known to have a major role in carcinogenesis.24 Therefore, targeting therapies based on mTOR inhibition have been developed for several types of human cancer. The analysis of a murine model that combined ablation of the B-cell receptor and activation of specific downstream signaling cascades suggested that antigen receptor–deficient mature B cells could be rescued by activated PI3K-α.25 Additional studies in murine models have indicated that PI3K signaling and FOXO transcription factors have opposing roles at several stages in the development of GC B cells. FOXO1 is known to regulate the expression of the gene coding for activation-induced cytidine deaminase (AID), among others. Abundant nuclear FOXO1 expression was found in the GC dark zone (the proliferative compartment), whereas upregulation of PI3K activity and downregulation of FOXO1 was observed in the GC light zone (the compartment in which cells are selected for further differentiation).26 Somatic hypermutation and proliferation were maintained in murine GC B cells in which PI3K-α activity was artificially induced, whereas class switch recombination was partly lost through a failure of switch-region targeting by AID.26 The analysis of a murine model in which the common pathogenic GOF mutation was introduced into PIK3CD further highlighted an intrinsic B-cell defect in class-switch recombination and suggested that impaired induction of AID was responsible.27 Similar observations have been made in APDS patients.9,10,27
However, it is still not clear to what extent disturbed expression and/or off targeting of AID (relative to increased activation of the PI3K pathway in B cells) contributes to the occurrence of B-cell lymphoma in APDS.
Furthermore, PI3K-δ has an important role in several T lymphocyte functions. A study of a knock-in mouse model carrying p110δ kinase-dead mutations highlighted a possible role for p110δ-mediated PI3K activity in TFH cells within the GC. TFH cell activity controls B-cell proliferation and modulates the magnitude of the GC reaction through inducible costimulator–regulated p110δ-mediated PI3K signaling, which regulates the production of key TFH cytokines and effector molecules like interleukin-21.28,29 Several independent studies have observed an elevated circulating TFH cell count in patients with APDS.30-32 An analysis of the murine model of APDS1 also highlighted aberrant TFH cell function.32 An abnormally high TFH cell count has also be observed in APDS1 and APDS2 lymphoid organs, including massive hyperplasia of PD-1+ TFH cells in the GCs.14,15,31 It has been hypothesized the disruption of the interaction between p85α and mutated p110δ causes the former to interact more strongly with osteopontin. This interaction has been shown to promote TFH cell differentiation in mice.33 The abnormally high TFH cell count is reminiscent of descriptions of the microenvironment in B-cell lymphomas (especially follicular lymphoma).34 The disturbance of TFH cell function might therefore contribute to the elevated frequency of B-cell lymphoma in APDS.
Another possible promalignancy factor in APDS is the impairment in tumor immune surveillance caused by defective T cell– and NK cell–mediated cytotoxicity. The premature immunosenescence/exhaustion phenotype of CD8+(CD57+) T cells and changes in NK-cell differentiation are both involved in this impairment in cytotoxicity.35 Moreover, an impairment of p110δ activity in regulatory T cells (Tregs) has been shown to break tumor-induced immune tolerance in murine models.36 The functional importance of PI3K-δ–mediated signaling in Tregs has also been shown in a study of knock-in mice carrying p110δ kinase-dead mutations.37 Patients with APDS are variously reported as having low, normal, or high circulating Treg counts, further highlighting the immunophenotypic heterogeneity of this disorder.9,31,32,38 It is clear that further investigation of Treg function in APDS is required.
Taken as a whole, these findings indicate that uncontrolled proliferation and malignant transformation of B lymphocytes in patients with APDS are due to B cell–intrinsic and B cell–extrinsic mechanisms (ie, elevated TFH cell counts, impaired cytotoxicity in CD8 T and NK cells, and defects in Treg activity).
Somatic PIK3CD and PIK3R1 mutations in cancer
The involvement of PI3K in carcinogenesis is further suggested by the detection of somatic GOF mutations in PIK3CD and PIK3R1 in several cancer tissues; these mutations highlight a potential oncogenic role for hyperactive PI3K in the development of neoplasia.
The exact same somatic PIK3CD GOF mutations found in APDS1 patients (eg, c.3061G>A; p.E1021K) have been described in patients with various lymphoid malignancies, such as DLBCL,39 acute lymphoblastic T-cell leukemia,40 hepatosplenic T-cell lymphoma,41 mantle cell lymphoma,42 and (more rarely) Burkitt lymphoma,43 as well as in nonlymphoid malignancies like malignant melanoma.44 Somatic PIK3R1 mutations have been described in Burkitt lymphoma45 and are also prevalent in endometrial carcinoma, metastatic prostate carcinoma, glioblastoma, and colorectal adenocarcinoma.46 This broad pattern of nonlymphoid malignancies reflects the ubiquitous expression of p85α and indicates the importance of the interaction of PIK3R1-encoded regulatory subunits with the ubiquitously expressed catalytic subunit p110 (eg, p110α) as well as with phosphatase and tensin homolog.47
Interestingly, the splice site mutations in APDS2 have also been described as somatic mutations in nonlymphoid malignancies (including breast cancer)48,49 ; this indicates that the shortened p85α protein has harmful effects in cells other than leukocytes. To our knowledge, there is only 1 report of an APDS2 patient with breast cancer.15 Surprisingly, the splice site mutations that cause APDS2 have (again, to our knowledge) not yet been reported as somatic mutations in B-cell lymphoma cohorts (eg, Burkitt lymphoma, CHL, or DLBCL). This disparity might be due to the focus of the cohort studies on somatic mutations in coding regions.
Treatment
The treatment of APDS is primarily symptomatic and is based on preventing or fighting infections with prophylactic or curative antibiotic therapy, antiviral drugs, and immunoglobulin replacement therapy. Steroids and anti–B-cell monoclonal antibodies can be useful in cases of severe autoimmunity or lymphoproliferation. Given that cancer in general and lymphoma in particular are the major complications of APDS, a focus on early diagnosis and effective treatment is essential. Physicians should pay careful attention to lymphoproliferation, a frequent complication that impairs a patient’s quality of life. Surgery is then required (often iteratively), with excision of the spleen, lymph nodes, tonsils, and adenoids. A precise histologic assessment is essential, with the evaluation of proliferation markers and monoclonality. Although the differential diagnosis is sometimes challenging, it is essential to distinguish between an EBV-induced polymorphic B lymphoproliferative syndrome and an actual lymphoma.50 The former can be controlled by treatment with an anti–B-cell monoclonal antibody, whereas the latter requires aggressive chemotherapy and (in some cases) radiotherapy or even autologous hematopoietic stem cell transplantation (HSCT).
Along with symptomatic therapies, etiologic (disease curative) treatment approaches aimed at controlling PI3K, sometimes even before APDS is diagnosed, have been developed. Allogeneic HSCT has been reported in the literature and seems to be associated with good clinical outcomes.51,52 Rapamycin (which inhibits the mTOR pathway downstream of PI3K signaling; Figure 1) is effective in patients with lymphoproliferation but has harmful adverse effects (strong immunosuppression and kidney and liver failure).22,53 The treatment of APDS patients with specific p110δ inhibitors is an attractive concept, because the latter compounds have been found to reduce PI3K-δ activity in cell lines carrying mutations in p110δ and p85α.9,11,12,54 The proven efficacy and acceptable safety profile of the PI3K-δ inhibitor idelalisib in the treatment of patients with CLL55,56 and patients with indolent non-Hodgkin lymphoma who had previously received a number of other treatments57 further argue in favor of this kind of therapy. Nevertheless, the administration of idelalisib to patients with relapsed indolent non-Hodgkin lymphoma was associated with various adverse events (colitis, pneumonitis, diarrhea, neutropenia, and elevated serum alanine or aspartate aminotransferase levels).55-57 Furthermore, the potential for genomic instability in B cells (via the increased expression and off-target activity of AID) must to be taken into consideration when administering p110δ-specific inhibitors.58 Several clinical trials of the efficacy and safety of inhaled or oral p110δ-specific inhibitors in APDS patients are ongoing (registered at www.clinicaltrials.gov as #NCT02435173 and #NCT02593539 and at https://eudract.ema.europa.eu as #2015-002900-10).59 The preliminary data from the NCT02435173 trial (using the oral PI3K-δ inhibitor leniolisib) are encouraging, and no significant adverse events have been reported so far.60 It has not yet been shown that p110δ-specific targeted therapy can prevent lymphoma or other cancers, although this would be expected if PI3K activity and immune surveillance were normalized (despite a possible effect on AID function in B cells). Long-term follow-up studies of the efficiency and safety of p110δ-specific inhibitors are needed, especially because lifelong treatment would be required.
At present, it is still not clear whether potentially curative HSCT (especially when an HLA-identical donor is available) should be preferred to drug treatment. The marked and unexplained heterogeneity of the clinical manifestations in APDS patients (even within the same family) prevents formal recommendations from being draw up. Lastly, the identification of prognostic biomarkers would be of value in choosing appropriate treatments and follow-up procedures.
Discussion
Activated PI3K-δ syndrome is a frequent cause of autosomal dominant primary combined immunodeficiency. Along with recurrent infections and autoimmunity (ie, common features of combined immunodeficiencies in general), lymphoproliferation is a clinical hallmark of this syndrome. Cancer (mainly B-cell lymphoma) is the main complication and is associated with a much worse prognosis. The accurate, early diagnosis of APDS on the basis of a patient’s clinical characteristics alone is challenging because of the great phenotypic heterogeneity. In fact, B-cell lymphoma can even be the revealing symptom in APDS. An analysis of PI3K signaling (the phosphorylation status of the downstream targets AKT or ribosomal protein S6) might be useful, although sequencing of the PIK3CD and PIK3R1 genes is currently the only means of obtaining an unambiguous diagnosis. It is noteworthy that in contrast to preliminary indications of E1021K as a hotspot in the PIK3CD gene, a range of GOF mutations in different domains have now been described; therefore, sequencing of the whole gene is required.61,62 A detailed assessment at diagnosis is required for appropriate genetic counseling, treatment, and follow-up with a view toward the early detection of any malignancies. Allogeneic HSCT and PI3K-δ or mTOR inhibition signaling are treatment options worth considering and might prevent the development of cancer.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by INSERM, the Agence National de la Recherche (as part of the Investment for the Future program by ANR-10-IAHU-01 and by ANR-15-CE15-0020 [ANR-PIKimun]), the Ligue Contre le Cancer–Comité de Paris, the Fondation ARC pour la recherche sur le Cancer, and the Centre de Référence Déficits Immunitaires Héréditaires. S.K. is a Centre National de la Recherche Scientifique staff researcher.
Authorship
Contribution: A.D. and S.K. analyzed data and wrote the manuscript.
Conflict-of-interest disclosure: The Laboratory of Human Lymphohematopoiesis received fees for services from UCB Biopharma SPRL. S.K. is an inventor of the International patent “A specific trifluoroethyl quinoline analogue for use in the treatment of APDS,” WO 2017/198590. A.D. declares no competing financial interests.
Correspondence: Anne Durandy, Institut Imagine, INSERM U1163, 24 Boulevard du Montparnasse, 75015 Paris, France; e-mail: anne.durandy@inserm.fr.
