


Abstract
The clinical development of effective cancer immunotherapies, along with advances in genomic analysis, has led to the identification of tumor environmental features that predict for sensitivity to immune checkpoint blockade therapy (CBT). Early-phase clinical trial results have demonstrated the remarkable effectiveness of CBT in specific lymphoma subtypes, including classical Hodgkin lymphoma and primary mediastinal B-cell lymphoma. Conversely, CBT has been relatively disappointing in follicular lymphoma and diffuse large B-cell lymphoma. These clinical observations, coupled with important scientific discoveries, have uncovered salient features of the lymphoma microenvironment that correlate with immunotherapy response in patients. For example, classical Hodgkin lymphoma is characterized by an inflammatory environment, genetic alterations that facilitate escape from immune attack, and sensitivity to PD-1 blockade therapy. On the other hand, for lymphomas in which measures of immune surveillance are lacking, including follicular lymphoma and most diffuse large B-cell lymphomas, anti-PD-1 therapy has been less effective. An improved understanding of the immune landscapes of these lymphomas is needed to define subsets that might benefit from CBT. In this article, we describe the immune environments associated with major B-cell lymphomas with an emphasis on the immune escape pathways orchestrated by these diseases. We also discuss how oncogenic alterations in lymphoma cells may affect the cellular composition of the immune environment and ultimately, vulnerability to CBT. Finally, we highlight key areas for future investigation, including the need for the development of biomarkers that predict for sensitivity to CBT in lymphoma patients.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 590.
Disclosures
Author Justin Kline has received research support from Merck, iTeos, and Verastem; served on advisory boards sponsored by Merck, Kite/Gilead, Seattle Genetics, and Verastem; and is a member of a speakers' bureau for Kite/Gilead. Stephen M. Ansell has received research support from Bristol-Myers Squibb, Seattle Genetics, Regeneron, Pfizer, Affimed, Trillium, AI Therapeutics, and Takeda. Associate Editor Freda K. Stevenson, CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, and author James Godfrey declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe immune environments, immune escape pathways, and oncogenic changes associated with "inflamed" subtypes of major B-cell lymphomas, according to a review
Determine immune environments, immune escape pathways, and oncogenic changes associated with "noninflamed" subtypes of major B-cell lymphomas, according to a review
Identify response to immune checkpoint blockade therapy in "inflamed" vs "noninflamed" subtypes of major B-cell lymphomas, according to a review
Release date: February 20, 2020; Expiration date: February 20, 2021
Introduction
Immunotherapy with monoclonal antibodies that interfere with adaptive immune checkpoints has revolutionized the treatment of numerous human cancers.1,2 In particular, therapies that inhibit interactions between PD-1+, tumor-reactive T cells, and PD-L1–expressing malignant or immune cells in the local environment have been remarkably effective in patients with select solid tumors and lymphomas. Recent studies have identified characteristics of tumor environments that are associated with sensitivity to immune checkpoint blockade therapy (CBT),3-6 and have found that antitumor immune responses are strongly influenced by the activation of oncogenic pathways in malignant cells.7-10 Collectively, this knowledge has emphasized the importance of a preexisting antitumor immune response in predicting clinical benefit from CBT.
In lymphomas, the importance of the immune environment in regulating the efficacy of CBT is increasingly being recognized. For instance, in response to potent host immune surveillance, Hodgkin-Reed-Sternberg (HRS) cells acquire highly recurrent copy gains of the chromosomal region containing the PD-L1 locus,11-15 implicating PD-L1 upregulation as a key immune escape mechanism in classical Hodgkin lymphoma (cHL),16,17 which is exquisitely sensitive to PD-1 blockade therapy.18-22 Conversely, PD-L1 copy gains are not observed in follicular lymphoma (FL) or in most diffuse large B-cell lymphoma (DLBCL),23,24 which are typically resistant to anti–PD-1 therapy.25,26 Thus, PD-L1 gene alterations identify lymphomas against which host immune responses have been generated, and their presence may serve as a predictive biomarker of response to PD-1 blockade therapy.24
In this review, we discuss current knowledge regarding the immune landscapes associated with major B-cell lymphoma subtypes and distinguish “inflamed” from “noninflamed” lymphomas to provide a framework for conceptualizing how particular immune environments promote vulnerability or resistance to CBT.
Immune landscapes in lymphoma: inflamed vs noninflamed phenotypes
Advances in the characterization of immune landscapes in solid tumors have led to the notion that cancers can be broadly divided into those exhibiting T-cell inflamed or T-cell noninflamed phenotypes.2,4,27 T-cell inflamed tumors are characterized morphologically by robust immune cell infiltration and transcriptionally by the upregulation of genes expressed in activated T cells and those induced downstream of interferon-γ (IFN-γ) signaling.3,6 Importantly, T-cell inflamed tumors are also enriched for sensitivity to CBT.3,6 Conversely, T-cell noninflamed tumors are largely devoid of infiltrating immune cells, and are typically resistant to CBT.2
Extrapolating the concept of the inflamed vs noninflamed phenotypes to lymphomas has been complicated because most originate in secondary lymphoid organs where immune cells normally reside. Nonetheless, genomic studies and observations from CBT clinical trials have identified lymphoma subtypes commonly associated with host immune responses, as well as those that are not. Shared characteristics of inflamed lymphomas include the presence of a prominent immune cell infiltrate that notably includes T cells,28,29 acquisition of recurrent genetic alterations that facilitate escape from immune surveillance,11-15,23,24,30-33 and frequent mutations resulting in constitutive NF-κB pathway activation (Figures 1 and 2A).24,31,34-36 Epstein-Barr virus (EBV) infection, known to be involved in the pathogenesis of several lymphoma subtypes, including cHL and rare DLBCLs, may also promote an inflamed environment and provide a source of foreign antigens for host T-cell recognition (Figure 2B).37 Some inflamed lymphomas, including subsets of cHL and primary mediastinal large B-cell lymphoma (PMBL), also harbor genetic alterations leading to defective DNA mismatch repair and microsatellite instability, as well as exhibit apolipoprotein B messenger RNA (mRNA) editing enzyme, catalytic polypeptide-like (APOBEC) mutational signatures (Figure 2B),38 both of which have been linked to anti–PD-1 therapy response in solid tumors because of their association with increased mutational load and neo-antigen generation.39,40 Finally, inflamed lymphomas tend to be sensitive to PD-1 blockade therapy.18-22,24
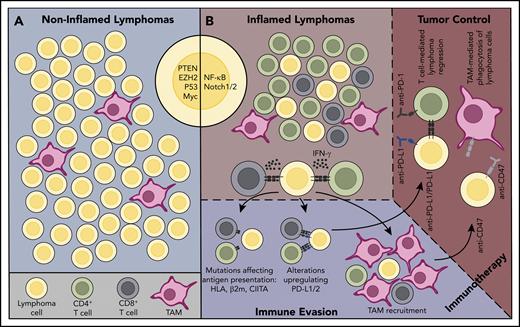
Inflamed and noninflamed lymphoma environments. (A) A noninflamed lymphoma environment. This lymphoma lacks infiltration by T cells. Oncogenic alterations that may contribute to poor immune cell infiltration/activation in noninflamed lymphomas are shown (PTEN, EZH2, TP53, MYC). Because of the lack of immune recognition, genetic alterations associated with escape from immune surveillance are typically lacking. This lymphoma would be predicted to be resistant to immune CBT therapy with anti–PD-1 or anti–PD-L1 antibodies. (B) An inflamed lymphoma environment. This lymphoma environment is enriched in infiltrating immune cells, including CD4+ and CD8+ T cells. Oncogenic signaling pathways that may promote an inflammatory lymphoma environment, such as NF-κB and NOTCH, are shown. In response to selective pressure from an ongoing antilymphoma immune response, the lymphoma cells have acquired genetic alterations that enhance expression of PD-L1/2 and that lead to defective antigen presentation to T cells. This lymphoma has also recruited numerous TAMs. As such, this lymphoma would be expected to be susceptible to immunotherapy with PD-1 or PD-L1 blockade therapy, and might also be more responsive to immunotherapy with antibodies that block CD47/SIRPα interactions.
Inflamed and noninflamed lymphoma environments. (A) A noninflamed lymphoma environment. This lymphoma lacks infiltration by T cells. Oncogenic alterations that may contribute to poor immune cell infiltration/activation in noninflamed lymphomas are shown (PTEN, EZH2, TP53, MYC). Because of the lack of immune recognition, genetic alterations associated with escape from immune surveillance are typically lacking. This lymphoma would be predicted to be resistant to immune CBT therapy with anti–PD-1 or anti–PD-L1 antibodies. (B) An inflamed lymphoma environment. This lymphoma environment is enriched in infiltrating immune cells, including CD4+ and CD8+ T cells. Oncogenic signaling pathways that may promote an inflammatory lymphoma environment, such as NF-κB and NOTCH, are shown. In response to selective pressure from an ongoing antilymphoma immune response, the lymphoma cells have acquired genetic alterations that enhance expression of PD-L1/2 and that lead to defective antigen presentation to T cells. This lymphoma has also recruited numerous TAMs. As such, this lymphoma would be expected to be susceptible to immunotherapy with PD-1 or PD-L1 blockade therapy, and might also be more responsive to immunotherapy with antibodies that block CD47/SIRPα interactions.
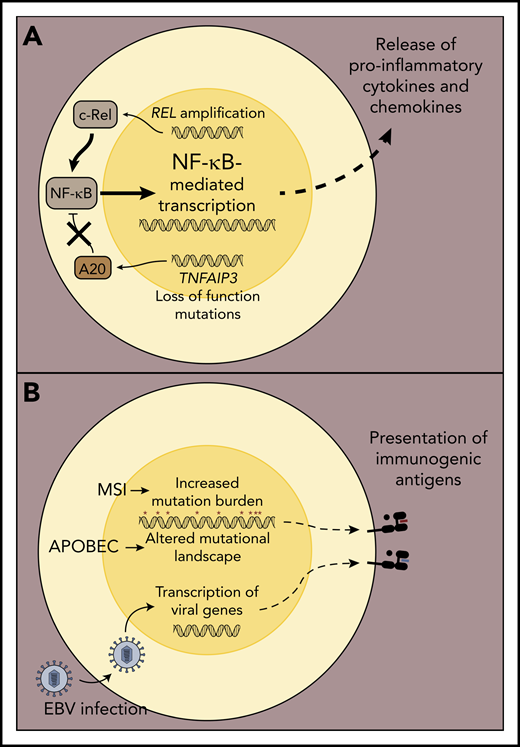
Mechanisms associated with inflamed lymphoma environments. (A) Acquisition of oncogenic alterations that drive increased expression of NF-κB pathway members or those resulting in the deletion or inactivation of NF-κB regulatory genes culminate in enhanced lymphoma cell-intrinsic NF-κB signaling and transcription of pro-survival genes, as well as those encoding pro-inflammatory cytokines and chemokines. The latter may lead to enhanced recruitment and sustained activation of immune cells in the lymphoma environment. (B) A subset of inflamed lymphomas, including cHL and PMBL, exhibit microsatellite instability and/or apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) mutational signatures, which have been associated with increased generation of neo-antigens that can be targeted by host T cells. Alternatively, malignant cells in some cHL and DLBCL are associated with EBV infection, and EBV-derived viral epitopes can drive T-cell responses, particularly in the context of cHL.
Mechanisms associated with inflamed lymphoma environments. (A) Acquisition of oncogenic alterations that drive increased expression of NF-κB pathway members or those resulting in the deletion or inactivation of NF-κB regulatory genes culminate in enhanced lymphoma cell-intrinsic NF-κB signaling and transcription of pro-survival genes, as well as those encoding pro-inflammatory cytokines and chemokines. The latter may lead to enhanced recruitment and sustained activation of immune cells in the lymphoma environment. (B) A subset of inflamed lymphomas, including cHL and PMBL, exhibit microsatellite instability and/or apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) mutational signatures, which have been associated with increased generation of neo-antigens that can be targeted by host T cells. Alternatively, malignant cells in some cHL and DLBCL are associated with EBV infection, and EBV-derived viral epitopes can drive T-cell responses, particularly in the context of cHL.
Responsiveness to anti–PD-1 therapy reveals much about previous interactions between a lymphoma and the host immune system because PD-1 blockade therapy is thought to function primarily by reactivating existing antitumor T-cell responses in situ, rather than through triggering de novo antitumor T-cell priming.41,42 Thus, CBT-responsive lymphomas (ie, inflamed lymphomas) are associated with ongoing, yet suppressed antilymphoma immune responses that can be effectively reinvigorated through CBT. In contrast, infiltrating immune cells, specifically T cells, are less conspicuous in noninflamed lymphomas, either because of the activation of lymphoma cell–intrinsic transcriptional programs that promote immune ignorance (ie, through downregulation of antigen presentation machinery) or that prevent the effective recruitment of immune cells into the tumor environment (Figure 3). Furthermore, noninflamed lymphomas typically lack genetic immune escape alterations and are often resistant to anti–PD-1 therapy.25,26 In this review, we operationally classify lymphomas as either inflamed or noninflamed based on the general characteristics outlined above. However, a large degree of heterogeneity exists in the actual immune environments that accompany lymphomas of the same type, as well as within those of differing histologies.
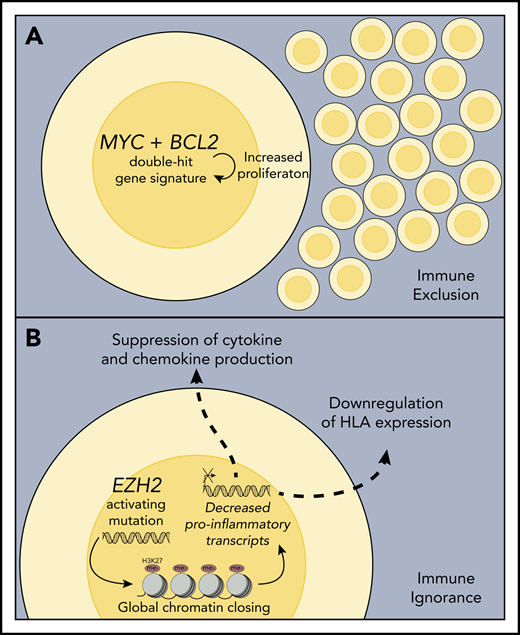
Mechanisms associated with noninflamed lymphoma environments. (A) Double-hit lymphomas or those expressing a so-called double-hit gene signature exhibit strong cell autonomous growth and survival programs and are often characterized by an intrinsically high proliferation rate, which may function to exclude immune cells from entering the lymphoma environment, thereby preventing the generation of a host antilymphoma immune response. (B) Activating EZH2 mutations lead to increased deposition of repressive histone marks (H3K27me3) on genes involved in B-cell differentiation, expression of HLA class I and class II molecules, and expression of chemokines required to recruit activated T cells to the lymphoma environment, rendering the host immune system ignorant to the presence of these tumors.
Mechanisms associated with noninflamed lymphoma environments. (A) Double-hit lymphomas or those expressing a so-called double-hit gene signature exhibit strong cell autonomous growth and survival programs and are often characterized by an intrinsically high proliferation rate, which may function to exclude immune cells from entering the lymphoma environment, thereby preventing the generation of a host antilymphoma immune response. (B) Activating EZH2 mutations lead to increased deposition of repressive histone marks (H3K27me3) on genes involved in B-cell differentiation, expression of HLA class I and class II molecules, and expression of chemokines required to recruit activated T cells to the lymphoma environment, rendering the host immune system ignorant to the presence of these tumors.
Inflamed lymphomas
cHL is the prototypical inflamed lymphoma, in which malignant HRS cells are heavily outnumbered by infiltrating immune cells. HRS cells secrete chemokines that recruit a variety of immune cells, the functions of which are coopted to create an environment supportive of HRS cell survival and growth.29 Conventional CD4+ T cells are more conspicuous than CD8+ T cells in cHL specimens43,44 and are enriched for expression of classical TH1 transcription factors (T-BET), cytokines (IFN-γ), and chemokine receptors (CXCR3, CCR5).44,45 This TH1-skewed phenotype might be expected to drive effective immunity, but the immune escape mechanisms activated by HRS cells subvert the effector functions of lymphoma-specific T cells and promote disease progression.29,46 The remarkable activity of PD-1 blockade therapy in relapsed/refractory (r/r) cHL,18-20,22 however, suggests that this dysfunctional T-cell phenotype is reversible when dominant inhibitory immune checkpoints are disabled.
PMBL shares biological and clinical features with the nodular-sclerosis variant of cHL.16,17,47 Little is known about the nature of the immune cell infiltrate in PMBL tumors. However, the observations that PMBL cells acquire recurrent genetic immune escape mechanisms,12,15,32,48,49 and that PMBL is highly responsive to anti–PD-1 therapy,21 suggest the presence of a baseline antilymphoma immune response in this disease, and support its categorization as an inflamed lymphoma subtype. Clearly, new research is needed to better understand the immune environment associated with PMBL.
In comparison with cHL, the immune landscape in DLBCL is more heterogeneous. Although a proportion of DLBCLs are capable of stimulating a host immune response as evidenced by expression of immune-related gene signatures (the so-called host response cluster) and acquisition of genetic alterations associated with immune evasion,23,24,33,50 most can be classified as noninflamed, which likely explains the modest efficacy of anti–PD-1 therapy in unselected r/r DLBCL patients.26
Immune escape mechanisms in inflamed lymphomas
A prominent feature of inflamed lymphomas is their ability to activate mechanisms that facilitate immune escape (Figure 1), a property initially described in solid cancers accompanied by vigorous antitumor immune responses.51 These include recurrent genetic alterations that drive PD-L1 upregulation and promote defective antigen presentation. This group of lymphomas is also adept at recruiting immune suppressive cell populations, including tumor-associated macrophages (TAM). Key immune escape pathways used by inflamed lymphomas are discussed next.
Genetic mechanisms that drive PD-L1 expression
Classically, PD-L1 upregulation on cancer cells and other cells in the tumor environment is mediated by IFN-γ produced locally by effector T cells.1 This adaptive mechanism of PD-L1 upregulation is common in solid cancers and generally indicates the presence of an antitumor T-cell response.1,52 In contrast, upregulated expression of the PD-1 ligands in lymphoma cells is often driven by genetic alterations, including structural variations (SVs) in the chromosomal region containing the PD-L1 and PD-L2 loci,12,14,15,23,24 or within the 3′ untranslated region (UTR) of the PD-L1 gene.53
Recurrent copy gains of the chromosome 9p24.1 region were first recognized in PMBL and cHL in the 1990s.48,54,55 Analysis of copy number and transcriptional data from cHL and PMBL cell lines subsequently identified PD-L1 and PD-L2 as key constituents of the 9p24.1 amplicon, suggesting their involvement in the pathogenesis of these related lymphomas.12 Quantitative polymerase chain reaction–based DNA copy number analysis of laser capture microdissected HRS cells or primary PMBL specimens revealed 9p24.1 SVs in 38% and 63% of cHL and PMBL samples, respectively. Chromosome 9p24.1 SVs were strongly correlated with increased PD-L1 and/or PD-L2 protein expression on cHL and PMBL cell lines and in primary samples.12 JAK2, also contained within the 9p24.1 amplicon, was shown to further augment PD-L1/2 expression in cHL cells.12 Chromosomal rearrangements involving PD-L1 and PD-L2, also resulting in enhanced expression, are particularly prominent in PMBL.15
A more recent study identified the presence of PD-L1 SVs in nearly all diagnostic cHL samples examined via fluorescence in situ hybridization (FISH).14 Within and across cHL specimens, a spectrum of PD-L1 gene alterations was observed, and these were classified according to the highest degree alteration present (amplification > relative copy gain > chromosome 9 polysomy). cHLs harboring overt PD-L1 gene amplifications expressed high levels of PD-L1 protein and contained few residual PD-L1 disomic HRS cells, likely a consequence of exposure to intense T-cell surveillance and selective outgrowth of highly PD-L1 gene-amplified HRS clones. PD-L1 amplifications were enriched in patients with advanced-stage disease and correlated with inferior progression-free survival (PFS) following initial treatment.14
Interestingly, although PD-L1 SVs were found in both EBV− and EBV+ cHLs, EBV+ cases tended to exhibit higher PD-L1 protein expression. Furthermore, upregulated PD-L1 expression was seen in a proportion of PD-L1 disomic cHLs.14 Together, these observations suggested that alternative mechanisms of PD-L1 upregulation were operational. Constitutive expression of the AP-1 heterodimeric proteins, cJUN and JUNB, had previously been demonstrated in HRS cells,56,57 and a highly conserved enhancer element containing tandem AP-1 sites was identified within intron 1 of the PD-L1 locus, capable of driving PD-L1 expression.13 Furthermore, in EBV-transformed lymphoblastoid cell lines, the AP-1 responsive enhancer element in PD-L1 was active, and the EBV-encoded latent membrane protein 1 led to enhanced cJUN activity. Latent membrane protein 1 was also found to induce direct activation of the PD-L1 promoter in a JAK/STAT-dependent manner.13 These results uncovered novel mechanisms of PD-L1 upregulation in cHLs lacking 9p24.1 alterations and in EBV-associated lymphomas.
PD-L1 upregulation occurs more sporadically in DLBCL. A large, immunohistochemistry-based analysis identified PD-L1 expression on lymphoma cells in 11% of DLBCLs analyzed. PD-L1 upregulation was more common in non–germinal center B-cell (GCB) and EBV+ DLBCLs.58 Using PD-L1 FISH, we and others have identified PD-L1 SVs in ∼20% to 25% of DLBCLs, with a strong enrichment in non–GCB-like cases.23,24 We showed that the types and penetrance (ie, the fraction of PD-L1 gene-altered lymphoma cells over the total population of lymphoma cells analyzed) of PD-L1 SVs varied widely among DLBCL specimens. Relative PD-L1 copy gains were most prevalent, but PD-L1 amplifications, chromosome 9 polysomy, and translocations involving PD-L1 were also found.24 PD-L1 protein expression was highest in DLBCLs with PD-L1 amplifications and translocations and was rare in those lacking PD-L1 SVs Similar to observations in cHL, DLBCLs with high-degree PD-L1 SVs (amplifications and translocations) contained few residual PD-L1 disomic lymphoma cells.24 PD-L1 SVs have also been observed in rare, extranodal DLBCLs including primary central nervous system lymphoma (PCNSL) and primary testicular lymphoma (PTL),59 which is somewhat surprising given their association with “immune privileged” sites.
Another unique genetic driver of PD-L1 upregulation in DLBCL involves recurrent SVs in the 3′ UTR of the PD-L1 gene.53 Localized to a 3.1-kb breakpoint in the 3′ region of PD-L1, these SVs stabilize PD-L1 transcripts. presumably leading to increased PD-L1 protein translation.53 High “cytolytic” scores (mRNA levels of perforin and granzyme A) were observed in DLBCLs with PD-L1 3′ UTR SVs,5,53 suggesting that these genetic alterations, like other PD-L1 SVs, mark a subset of inflamed DLBCLs. Collectively, these observations reveal that PD-L1 upregulation in DLBCL, when it does occur, is commonly driven by genetic mechanisms.
Genetic alterations leading to defective antigen presentation
Display of tumor-derived peptide antigens on HLA class I and II molecules is required for cancer immune surveillance by CD8+ and CD4+ T cells, respectively. Loss of or decreased HLA expression is a well-recognized cancer immune escape mechanism,5,60,61 and numerous genetic alterations associated with class I and II HLA loss have been described in lymphomas.32,33,62,63 In cHL, for example, exome sequencing of purified HRS cells and targeted sequencing of cell-free circulating tumor DNA have revealed a high incidence of loss-of-function mutations in B2M,30,31 resulting in impaired cell-surface expression of the β2 microglobulin (β2M)/HLA class I complex. Similarly, breaks in the class II transactivator (CIITA) gene that lead to decreased expression of HLA class II genes are present in 15% of cHLs and 38% of PMBLs.32
By immunohistochemistry analysis, decreased or absent β2M and/or HLA class I protein expression was observed in 79% of cHL samples, whereas decreased or absent expression of HLA class II was identified in 67%.63 The striking prevalence of modulated HLA expression in cHL indicates a highly recurrent adaptive response by HRS cells to disrupt T-cell surveillance. Alternative mechanisms of dysregulated HLA expression, including epigenetic or posttranscriptional regulation of HLA genes and their products, have also been suggested to occur in cHL.
Genetic mechanisms leading to altered HLA expression, including B2M mutations/deletions,33 chromosome 6p21.32 deletions,62 and CIITA alterations,64 have also been recurrently observed in DLBCL. Nongenetic mechanisms of dysregulated cell-surface HLA expression, including cytoplasmic HLA trafficking, have also been reported in DLBCL.33 We observed that downregulated expression of HLA class I and II molecules was extremely common in DLBCLs with PD-L1 SVs, likely in response to enhanced immune surveillance.24 Furthermore, copy loss of 6p21.32 occurs in ∼50% of cases of PCNSL and PTL.59,62,65 Thus, genetic and other mechanisms that perturb antigen presentation are pervasive in lymphomas, particularly those with “inflamed” immune environments, and likely function in concert with PD-L1 SVs to promote immune escape.
Recruitment of immune suppressive macrophages
TAMs exert tumor-promoting and immunosuppressive functions and contribute to poor outcomes in numerous cancers.66-69 Although macrophages are conspicuous in a variety of lymphomas, their ability to promote immune suppression and influence outcomes is likely context dependent. Clinically, a gene expression signature characteristic of TAMs was enriched in diagnostic cHL specimens from patients who experienced primary treatment failure.70 Similarly, increased numbers of CD68+ macrophages in the cHL environment were associated with a higher likelihood of relapse and shorter disease-specific survival following autologous stem cell transplantation (ASCT).70 Thus, macrophages contribute to chemotherapeutic resistance in cHL.
Several putative immune suppressive mechanisms associated with TAMs have recently been identified in cHL. Elegant multiparameter immunofluorescence imaging revealed that the majority of PD-L1–expressing cells in the cHL environment were in fact macrophages, specifically those residing near HRS cells.71 Moreover, PD-L1+ macrophages were frequently in contact with PD-1+ CD4+ T cells, suggesting that macrophages drive CD4+ T-cell dysfunction via PD-1–PD-L1 interactions, and/or by preventing direct access to HRS cells.71 Perhaps combinations of PD-1 blockade with therapies aimed at depleting macrophages (anti-CSF1 antibodies), or those that promote an inflammatory macrophage phenotype (phosphatidylinositol 3-kinase-γ inhibitors), could effectively counteract the immune suppressive effects of TAMs in the cHL environment.
Studies performed in the pre-rituximab era also found associations between lymphoma-resident macrophages and poor outcomes in DLBCL.72 However, in the context of rituximab-based chemoimmunotherapy, TAMs and/or macrophage gene signatures appear to be associated with more favorable treatment outcomes,73,74 likely because of a requirement for macrophages in mediating antibody-dependent cellular phagocytosis of rituximab-targeted DLBCL cells.75,76
One major TAM function is the elimination of tumor cells via phagocytosis. Cancers activate pathways that enable them to escape this fate, including upregulating cell surface expression of an antiphagocytic protein called CD47 (integrin-associated protein).77 CD47 engagement with SIRPα activates a signaling cascade in the macrophage that inhibits myosin accumulation at the phagocytic interface.78 Chao et al first demonstrated enhanced CD47 expression on malignant B cells in a variety of lymphoma subtypes and observed that higher CD47 mRNA levels were associated with inferior outcomes in DLBCL patients treated with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone.79 Interestingly, blockade of CD47-SIRPα interactions enhanced macrophage phagocytosis of various B-cell lymphoma cell lines in vitro, an effect that was potentiated via provision of a second pro-phagocytic stimulus, such as an anti-CD20 antibody (rituximab). Furthermore, rituximab and CD47 blockade therapy were synergistic in mediating rejection of human lymphoma xenografts.79 Based on these results, clinical trials of CD47/SIRPα blockade therapy alone or in combination with antibodies that activate Fc-mediated phagocytosis are under way in r/r lymphomas. Early results have been quite promising,80 offering credence to the notion that targeting innate immune evasion pathways can be clinically efficacious.
Role of oncogenic alterations in promoting an inflamed lymphoma environment
Genetic alterations in oncogenes and/or tumor suppressor genes drive the malignant phenotype in cancer cells, but can also directly or indirectly affect the recruitment and activation of immune cells in the tumor environment.7-10,27 Alterations resulting in increased NF-κB signaling, for example, have been associated with an inflammatory environment in solid tumors.81,82 Similarly, a shared feature of inflamed lymphoma subtypes, such as cHL and PMBL, is the presence of recurrent genetic alterations associated with constitutive NF-κB activation.35,46,83,84 We have also recently demonstrated strong evidence of enhanced NF-κB activation in a subset of inflamed DLBCLs that harbor PD-L1 SVs,24 which was also observed in DLBCLs exhibiting a host response gene expression signature described by Monti et al.35,50 In addition to providing an intrinsic survival advantage, NF-κB activation may induce secretion of chemokines by lymphoma cells that result in enhanced immune cell recruitment (Figure 2A), as has been reported in a preclinical lung cancer model.85 At present, the link between lymphoma-intrinsic NF-κB activation and an inflamed environment is correlative. Furthermore, because the oncogenic alterations that underlie NF-κB activation30,31,34,36,83,84,86,87 and the upregulated expression of NF-κB target genes are not entirely overlapping in cHL, PMBL, and DLBCL,16,17,35 the contribution of NF-κB signaling to generating an inflamed environment likely varies between and even within lymphoma subtypes. For these reasons, it will be imperative to define the impact of oncogenic NF-κB signaling on antilymphoma immune responses in the context of preclinical lymphoma models.
Recent genomic analyses have uncovered additional oncogenic pathways that may contribute to generating an inflamed DLBCL environment. By analyzing whole exome sequencing data from 304 DLBCL samples, Chapuy et al identified 5 unique genetic clusters (C1-C5).34 Particularly relevant to this discussion are C1 DLBCLs. These lymphomas were characterized by recurrent BCL6 SVs, mutations in NOTCH2 pathway components, and FAS. C1 DLBCLs also carried frequent alterations in NF-κB pathway members, BCL10 and TNFAIP3. Alterations in genes important for immune surveillance were conspicuous in C1 DLBCLs, including inactivating mutations in B2M, FAS, and CD70, as well as recurrent PD-L1 SVs34 Thus, along with constitutive NF-κB activation, oncogenic NOTCH2 signaling may contribute to orchestrating an inflamed environment in DLBCL, although additional studies are needed to verify this hypothesis and to define underlying mechanisms. The finding that PD-L1 SVs and other genetic immune escape mutations occur sporadically in non-C1 DLBCLs argues that mutational clustering alone is insufficient to effectively differentiate inflamed from noninflamed DLBCLs.34 Furthermore, oncogenic alterations leading to constitutive NF-κB activation are quite heterogeneous in DLBCL, and the downstream effects on the local environment likely differ significantly. For example, C5 DLBCLs, enriched in CD79B and MYD88 mutations, known to activate NF-κB signaling,83 were largely devoid of genetic alterations associated with immune escape.34
A similar genomic analysis performed by Schmitz et al defined 4 genetic DLBCL clusters.36 Their so-called BN2 cluster, enriched for BCL6 fusions, along with recurrent NOTCH2, TNFAIP3, and BCL10 mutations, largely mirrored C1 DLBCLs described previously.34,36 Inactivating CD70 mutations were common among BN2 DLBCLs, again indicating that genetic immune escape mechanisms are enriched in lymphomas driven by oncogenic NF-κB and NOTCH2 signaling. A second, smaller DLBCL cluster (N1) containing NOTCH1 mutations most strongly expressed immune-related gene signatures, also compatible with an inflammatory microenvironment.36 Together, these studies have implicated oncogenic NOTCH signaling in regulating an inflammatory DLBCL environment. The degree to which NOTCH1/2 pathway mutations promote immune cell recruitment and/or activation in DLBCL is unknown, and requires confirmation.
Clinical efficacy and biomarkers of response to PD-1 blockade therapy in inflamed lymphomas
Phase 1/2 studies of the anti–PD-1 antibodies, nivolumab and pembrolizumab, reported high overall response rates (ORR) of 65% to 87% in r/r cHL patients.18-20,22 Partial responses were observed in most subjects, whereas complete responses (CR) were less common. PFS was impressive in patients that achieved CR, a subset of whom may never require additional therapy.19 However, for patients in partial response after anti–PD-1 therapy, median PFS was 12 to 15 months.19 The limited follow-up time of these studies precludes firm conclusions regarding the ability of anti–PD-1 therapy to produce long-term remissions. As a result, the timing of potentially definitive therapies (allogeneic stem cell transplantation) in patients responding to PD-1 blockade therapy continues to be challenging, and biomarkers predictive of durable responses to PD-1 blockade are needed. In the phase 2 Study of Nivolumab in Patients With Classical Hodgkin's Lymphoma (Registrational) (CheckMate 205) study of nivolumab, higher degree PD-L1 SVs, and high PD-L1 protein expression on HRS cells were associated with a best overall response of CR and prolonged PFS.20 Interestingly, HLA class II expression on HRS cells was associated with higher CR rates across all patients, and with prolonged PFS in patients with a longer interval between relapse from ASCT and nivolumab treatment.88 These candidate biomarkers require validation in prospective clinical trials.
In PMBL, PD-1 blockade therapy with pembrolizumab has also yielded excellent results, with an overall risk ratio (ORR) of 48%. In the phase 1b KEYNOTE-013 study, median duration of response and overall survival were not reached in the r/r PMBL cohort with 11.3 months of follow-up.21 The phase 2 KEYNOTE-170 study reported an ORR of 45% and a CR rate of 13% among 53 r/r PMBL patients. Median duration of response was not reached with a follow-up of 12.5 months, and 12-month PFS was 38%. In both studies, no patient who achieved CR relapsed during follow-up.89 As expected, PD-L1/2 SVs were common among evaluable biopsy specimens, and the presence of PD-L1 SVs (copy gains and amplifications) was associated with increased PD-L1 protein expression, which itself predicted for prolonged PFS in pembrolizumab-treated patients.89
Finally, anti–PD-1 therapy has also demonstrated impressive activity in small numbers of patients with r/r PCNSL and PTL, although no prospective clinical trials have been completed.90 The discovery of recurrent PD-L1 SVs in PCNSL and PTL was instrumental in providing the initial support for the use of PD-1 blockade in these rare, poor-prognosis DLBCLs.59
Noninflamed lymphomas
Although the majority of our discussion has been focused on aspects of inflamed lymphomas, most lymphomas harbor noninflamed environments, including many DLBCLs, FL, chronic lymphocytic leukemia, and Burkitt lymphoma. Features of noninflamed lymphomas include sparse infiltration by immune cells, a paucity of genetic immune escape alterations, and resistance to CBT (Figure 1). These lymphomas may also express molecular programs that prevent immune cells from entering the tumor environment,91 or may, through an inherently high proliferation rate, effectively exclude immune cells from the local environment and generating an antilymphoma response (Figure 3A). Thus, promoting immune “exclusion” or “ignorance” may be a common mechanism shared by some noninflamed lymphomas.
In DLBCL, the expression of immune-related gene signatures and acquisition of genetic immune evasion alterations suggests that some capable of triggering a host immune response.24,50 Most DLBCLs, however, lack these features, justifying their categorization as noninflamed lymphomas. Several lines of evidence have indicated that GCB-like DLBCLs, in particular, may be inherently predisposed toward a noninflamed phenotype. First, GCB-derived, high-grade B-cell lymphomas (HGBL) with MYC, BCL2, and/or BCL6 gene rearrangements, as well as DLBCL/HGBLs expressing a so-called double-hit gene expression signature, were shown to be relatively devoid of infiltrating T cells, lacked immune-related transcriptional signatures and mutations in NF-κB pathway genes, and contained few genetic alterations associated with immune escape.91 These lymphomas were enriched for EZH2-activating mutations and commonly showed absent or low-level expression of HLA molecules.91 This finding is interesting because EZH2 activation has been shown to promote HLA downregulation in several cancers, including DLBCL.92,93 Second, PD-L1 SVs, which mark a subset of inflamed DLBCLs, are rare in GCB-like DLBCLs,23,24 also suggesting that lymphomas arising from germinal center B cells either poorly activate host immune sensing mechanisms or express oncogenic programs that drive immune cell exclusion from the tumor environment.
Based on these observations, DLBCL/HGBL of GCB derivation may be particularly resistant to CBT, although exceptions may exist and should be sought. Despite their relative enrichment in markers of inflammation, most ABC-like DLBCLs also do not respond to CBT. Therefore, it will be critical to elucidate how noninflamed lymphoma immune environments arise so that strategies aimed at reversing the phenotype can be devised. For example, in DLBCLs that are “cold” because of a failure to trigger innate immune sensing mechanisms, one could envision inducing an inflammatory environment through administration of toll-like receptor or stimulator of interferon gene agonists, followed by anti–PD-1 antibody therapy.94 For DLBCLs in which oncogenic signaling pathways inhibit immune cell recruitment (discussed in the following section), targeted therapies could be used to enhance anti–PD-1 activity. Some lymphomas will be resistant to CBT in a way that cannot yet be understood or rectified, and here, CD19-directed CAR T-cell therapy may be the preferred immunotherapeutic option.95,96
Like DLBCL, the immune environment in FL is complex and poorly understood. Early gene expression profiling studies revealed an association between particular immune-related gene signatures in FL specimens and favorable long-term survival.97 Also, patient-specific, anti-idiotype vaccines have induced effective antilymphoma immunity associated with durable remissions in a subset of patients with FL.98 These observations suggest that a host immune response may be present in some FL patients. Conversely, PD-L1 gene alterations are rare in FL (J.G., unpublished observation, 5 December 2019), which is also generally insensitive to PD-1 blockade,25 calling into question the effect of immune surveillance in this disease. Additional studies of the FL environment are needed to determine whether a subset of inflamed FLs truly exists.
Role of oncogenic alterations in promoting the noninflamed lymphoma phenotype
Strong observational and mechanistic evidence has implicated specific oncogenic pathways in promoting a noninflamed phenotype in solid cancers, often through inhibiting immune cell recruitment and/or activation in the tumor.7-10 Data supporting oncogenic signaling in fostering a noninflamed immune environment in lymphoma are sparse. However, a recent genomic DLBCL analysis identified correlations between alterations in particular oncogenes/tumor suppressor genes and decreased expression of immune-related gene sets.87 Alterations in PTEN, EZH2, and TP53 were all associated with decreased expression of gene sets related to innate or adaptive immune cells and/or canonical functions associated with these cells. This is of particular interest because mutations in these genes have been linked with immune cell exclusion, dysfunction, or immune suppression in other cancers.8,99,100 Emerging data indicate that EZH2-activating mutations, in particular, are strongly associated with decreased HLA expression and T-cell infiltration in DLBCL.91,93 Figure 3B depicts mechanisms by which EZH2 activating mutations may impact the immune response in lymphomas. MYC has also been suggested to regulate the immune environment in preclinical models through inducing expression of immune checkpoint molecules (CD47, PD-L1),101 and via stimulating expression of chemokines that recruit TAMs and exclude T cells and other lymphocytes from the tumor environment.102 MYC is central to the biology of many lymphomas and may also be involved in creating noninflamed lymphoma environments as outlined previously.91 Clearly, additional studies are needed to better understand how oncogenic signaling contributes to generating cold immune environments in lymphomas. Defining oncogenic pathways that promote immune exclusion will be essential for the development of future studies testing PD-1 blockade with relevant targeted therapies in r/r lymphomas, including DLBCL, where effective treatments are needed.
Clinical results of PD-1 blockade therapy in noninflamed lymphomas
PD-1 blockade therapy has been disappointing to date in r/r DLBCL and FL. In a phase 1 study, nivolumab induced objective responses in 40% and 36% of patients with FL and DLBCL, respectively, although numbers of treated patients were small.25 A subsequent, large phase 2 study of nivolumab in r/r DLBCL reported a disappointing ORR of 10% among subjects treated following relapse from ASCT, and only 3% in ASCT-ineligible patients.26 Although the ORR was low, 31% of patients in the post-ASCT relapse cohort achieved disease stabilization or better, and several durable responses occurred. Objective responses were not clearly different between patients with GCB-like and non–GCB-like DLBCLs.26 FISH analysis revealed that low-level PD-L1 SVs were common in pretreatment biopsy specimens, but only 3% harbored overt PD-L1 gene amplifications. No clear correlation between PD-L1 SVs or PD-L1 protein expression and response to nivolumab was identified, although the number of responders was too low for meaningful statistical comparison.26 Conversely, in our retrospective analysis of the KEYNOTE-013 study, the presence of PD-L1 SVs was significantly associated with achievement of an objective response to pembrolizumab in r/r DLBCL patients.24 To prospectively define the utility of PD-L1 SVs as a predictive biomarker of response to PD-1 blockade, we have initiated a prospective phase 2 clinical trial of pembrolizumab in PD-L1 gene-altered r/r DLBCL (NCT03990961).
Unanswered questions and future directions
Over the past 5 years, we have witnessed the remarkable impact of anti–PD-1 therapy in r/r cHL and PMBL. Conversely, the future of CBT in r/r DLBCL remains unclear. It will be essential to gain a better understanding of the DLBCL immune environment through genomic and primary specimen analyses to identify disease subsets that may be vulnerable to anti–PD-1 therapy, and to develop predictive biomarkers for prospective evaluation in clinical trials. Furthermore, the degree to which immune checkpoints outside of PD-1/PD-L1 can be successfully targeted in patients with lymphoma needs to be defined, as does the utility of combining PD-1 blockade with conventional therapies in earlier lines of treatment.
In this review, we have proposed a subdivision of lymphomas into inflamed and noninflamed subsets based on environmental features and sensitivity to CBT. Operationally, this categorization is “black and white” and is based largely on the degree of observed responsiveness of lymphomas to CBT. In reality, lymphoma immune environments are highly heterogeneous and should be viewed as “shades of gray.” Regardless, this terminology provides a framework for conceptualizing lymphomas through a unique lens focused on clarifying underlying factors that regulate the lymphoma–immune interface. We envision that this perspective will also be useful in the development of future immunotherapeutic studies in lymphomas.
Acknowledgments
This research was supported by a V Foundation grant (T2018-008), a Leukemia & Lymphoma Society Grant (TRP-18189-19) (J.K.), an American Society of Hematology Research Training Award for Fellows, a Cancer Research Foundation Young Investigator Award, and a Sigal Immuno-oncology Fellowship (J.G.).
Authorship
Contribution: J.K., J.G., and S.M.A. conceived, drafted, and revised the manuscript.
Conflict-of-interest disclosure: J.K. has received research support from Merck, iTeos, and Verastem; served on advisory boards sponsored by Merck, Kite/Gilead, Seattle Genetics, and Verastem; and is a member of a speakers’ bureau for Kite/Gilead. S.M.A. has received research support from Bristol-Myers Squibb, Seattle Genetics, Regeneron, Pfizer, Affimed, Trillium, AI Therapeutics, and Takeda. J.G. declares no competing financial interests.
Correspondence: Justin Kline, 5841 S. Maryland Ave, MC 2115, University of Chicago, Chicago, IL 60637; e-mail: jkline@medicine.bsd.uchicago.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal