

Key Points
Intestinal GVHD in Atg16L1-deficient mice was reversed by inhibiting necroptosis.
An ex vivo platform incorporating organoids and T cells can recreate susceptibility to tissue injury and be applied to drug testing.
Visual Abstract

Abstract
A goal in precision medicine is to use patient-derived material to predict disease course and intervention outcomes. Here, we use mechanistic observations in a preclinical animal model to design an ex vivo platform that recreates genetic susceptibility to T-cell–mediated damage. Intestinal graft-versus-host disease (GVHD) is a life-threatening complication of allogeneic hematopoietic cell transplantation. We found that intestinal GVHD in mice deficient in Atg16L1, an autophagy gene that is polymorphic in humans, is reversed by inhibiting necroptosis. We further show that cocultured allogeneic T cells kill Atg16L1-mutant intestinal organoids from mice, which was associated with an aberrant epithelial interferon signature. Using this information, we demonstrate that pharmacologically inhibiting necroptosis or interferon signaling protects human organoids derived from individuals harboring a common ATG16L1 variant from allogeneic T-cell attack. Our study provides a roadmap for applying findings in animal models to individualized therapy that targets affected tissues.
Introduction
Treatment of complex inflammatory disorders often involves “step-up” approaches in which patients receive interventions of increasing intensity and risk after failure to demonstrate improvement with milder therapies. Multiple rounds of empiric testing and failure of treatments present a substantial burden on the health care system that contributes to decreased quality of life and can negatively impact the disease course. The promise of precision medicine is that certain features of the patient will predict responsiveness to therapies and circumvent the need for trial- and-error approaches. However, biomarker analysis of blood or other tissue specimens has had only limited success. An alternative approach is to establish an ex vivo assay in which disease-related events are recreated with patient-derived material and then subsequently applied to test drug responsiveness.
Allogeneic hematopoietic cell transplantation (allo-HCT) involving the transfer of bone marrow (BM), peripheral blood, or cord blood from a nonidentical donor can be a life-saving procedure. When applied to treat malignancies such as myeloid leukemia, donor-derived T cells contribute to remission by attacking tumor cells in recipients. In as many as 50% of transplant recipients, these alloreactive T cells attack healthy tissues to cause the multiorgan disorder graft-versus-host disease (GVHD).1 Damage to the gastrointestinal tract accounts for much of the morbidity and mortality associated with GVHD,2 yet few biomarkers and methods are currently available that predict intestinal involvement or response to treatment.3-5
We previously demonstrated that the autophagy gene ATG16L1 is protective during allo-HCT.6,7 A common ATG16L1 (ATG16L1T300A) variant was initially identified as a susceptibility factor for the inflammatory bowel disease (IBD) Crohn’s disease.8 Intestinal GVHD and Crohn’s disease frequently involve the distal small intestine but can involve any part of the gastrointestinal tract and are characterized by overproduction of T-helper 1 cytokines tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ), as well as epithelial barrier disruption.2,8,9 Based on these similarities, we examined the role of ATG16L1 in GVHD and found that mice with reduced Atg16L1 expression were susceptible to GVHD in an animal model of allo-HCT and that the ATG16L1T300A variant was associated with increased transplant-related mortality in human allo-HCT recipients.6 More recently, we showed that Atg16L1 deletion in intestinal epithelial cells (IECs) in mice is sufficient to confer increased lethality following allo-HCT.7
During autophagy, organelles and other cytosolic material are degraded and recycled when sequestrated by double-membrane vesicles that fuse with endolysosomes.10,11 Mice harboring IEC-specific deletions of ATG16L1 or other autophagy proteins display impaired viability of several epithelial lineages, including enterocytes, Paneth cells, and goblet cells.12-16 We and other investigators independently demonstrated that ATG16L1 and other autophagy components inhibit a form of programmed necrosis termed “necroptosis” in murine intestinal organoids,7,17,18 a 3-dimensional cell culture system in which IEC lineages are differentiated from epithelial stem cells.19 Necroptosis occurs when cytokine and death receptors induce the formation of a complex, consisting of receptor interacting protein kinase 3 (RIPK3) and RIPK1, that mediates the recruitment and phosphorylation of the pore-forming molecule mixed lineage kinase domain-like (MLKL).20 The role of autophagy proteins is cell type dependent and can promote necroptosis in prostate tumor cells.21-23 It is unclear how inhibiting ATG16L1 disrupts intracellular signaling to decrease the viability of IECs, and the relevance to human disease requires further investigation.
In this study, we apply a more clinically relevant mouse model of allo-HCT to demonstrate that intestinal GVHD in an Atg16L1-mutant setting can be ameliorated by blocking necroptosis signaling. We then develop an ex vivo platform, initially with samples from mice and then with human specimens, to recreate genetic susceptibility to T-cell–mediated damage by coculturing intestinal organoids with peripheral T cells. Our findings provide insight into how ATG16L1 protects against T-cell–mediated intestinal injury and establish a novel tool that enables the prediction of disease course and intervention outcomes.
Methods
Mice
Age- and sex-matched 6- to 15-week-old mice on the C57BL/6J (B6) background were used as recipients. Atg16L1f/f;villinCre (Atg16L1ΔIEC) and littermate control Atg16L1f/f mice were generated as previously described.7 f/f Ripk3−/− and ΔIEC Ripk3−/− mice were generated by crossing Atg16L1ΔIEC mice with Ripk3−/− mice, provided by Xiaodong Wang (National Institute of Biological Sciences). B6, B10.BR, and LP/J mice were purchased from The Jackson Laboratory and bred onsite to generate animals for experimentation. Atg4B−/− mice and Atg16L1T316A mice were previously described.7,24 All animal studies were performed according to approved protocols by the New York University School of Medicine and Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committees.
Human samples/study approval
Pinch biopsies were obtained with consent from adult IBD patients undergoing surveillance colonoscopy, using 2.8-mm standard biopsy forceps, after protocol review and approval by the New York University School of Medicine Institutional Review Board (Mucosal Immune Profiling in Patients with Inflammatory Bowel Disease; S12-01137). Inflammation status of tissue was confirmed by pathological examination.
For allogeneic T cells, peripheral blood mononuclear cells (PBMCs) from anonymous, healthy donors (New York Blood Center) were isolated by Ficoll gradient separation, as previously described.25 CD14+ monocytes were removed from the PBMC fraction by positive selection. The remaining negative fraction was used to isolate T cells. For syngeneic T cells, venous blood was collected at the time of endoscopic procedures in sodium heparin BD Vacutainer blood collection tubes (Becton Dickinson).
Statistical analysis
GraphPad Prism version 7 was used for statistical analysis. Differences between 2 groups were assessed by a 2-tailed unpaired Student t test when data were distributed normally. Analysis of variance with Tukey’s multiple-comparisons test was used to evaluate experiments involving multiple groups. Survival was analyzed with the Mantel-Cox log-rank test. Continuous variables in Figure 7A were analyzed by the Student t test, and categorical variables were analyzed by the χ2 test or Fisher’s exact test.
All other methods are described in detail in supplemental Information (available on the Blood Web site).
Results
ATG16L1 in IECs protects against GVHD mediated by RIPK1 and RIPK3
Previously, we found that mice with an IEC-specific deletion of Atg16L1 on the B6 background (Atg16L1ΔIEC mice) exhibited poor survival in an allo-HCT model in which recipients are irradiated and injected with BM and T cells from donor B10.BR mice (H-2k).7 Although this major histocompatibility complex (MHC)-disparate model was useful in identifying an IEC-intrinsic function of ATG16L1, the rapid onset that we observed suggests that this transplant procedure may not accurately reflect the course of GVHD in humans. Therefore, we examined whether the protective function of ATG16L1 can be detected in an improved MHC-matched allo-HCT model.26 Recipients were treated with busulfan and cyclophosphamide to mimic a chemotherapy-based conditioning regimen, which we confirmed depletes leukocytes, and injected with BM and T cells derived from LP/J mice (H-2b) (supplemental Figure 1A-B). Atg16L1ΔIEC recipient mice displayed 100% mortality and an increased disease score compared with the Cre-negative Atg16L1f/f control littermates, whereas all mice of both genotypes that received BM without T cells survived (Figure 1A-B). Thus, we validated previous findings; together, these 2 models show that Atg16L1 expression in IECs inhibits GVHD.
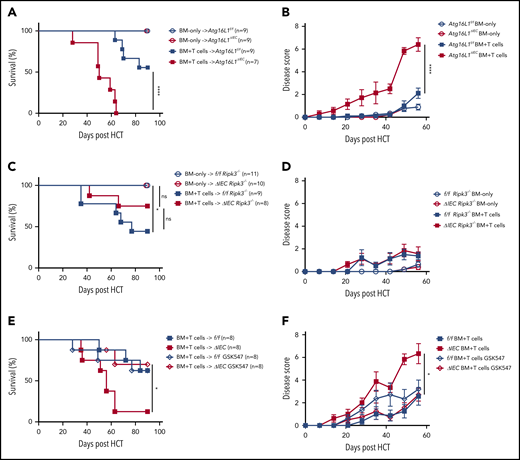
ATG16L1 in the intestinal epithelium protects against lethal GVHD mediated by RIPK1 and RIPK3. (A) Survival of Atg16L1f/f and Atg16L1ΔIEC mice receiving a chemotherapy conditioning regimen and transplanted with 5 × 106 T-cell–depleted BM cells, with or without 4 × 106 splenic T cells from donor LP/J mice. (B) Disease scores (see “Methods”) evaluated every 7 days after allo-HCT in (A). (C) Survival of chemotherapy-pretreated f/f Ripk3−/− and Atg16L1ΔIEC × Ripk3−/− (ΔIEC Ripk3−/−) mice transplanted with 5 × 106 T-cell–depleted BM cells, with or without 4 × 106 splenic T cells from donor LP/J mice. (D) Disease scores evaluated every 7 days after allo-HCT in (C). (E) Survival of chemotherapy-pretreated Atg16L1f/f (f/f) and Atg16L1ΔIEC (ΔIEC) mice that received GSK547 or control chow and were transplanted with 5 × 106 T-cell–depleted BM cells and 4 × 106 splenic T cells from donor LP/J mice. GSK547 was started 10 days before allo-HCT and continued until the end of the study. (F) Disease scores evaluated every 7 days after allo-HCT in (E). Data points in A, C, and E represent individual mice and are the combined results of 2 experiments performed independently. Data points in B, D, and F are mean disease scores of viable mice. Bars represent means ± standard error of the mean. For disease score, the area under the curve was determined for each mouse. *P < .05, ****P < .0001 analysis of variance with Tukey’s multiple-comparison test. HCT, hematopoietic cell transplantation; ns, not significant.
We next profiled immune parameters on day 28 after allo-HCT before the onset of lethality. We did not detect a significant effect of ATG16L1 deficiency on the amount of specific immune cells or cytokines, with the exception of a <twofold increase in IP-10 (CXCL10) (supplemental Figure 1C-D; supplemental Table 1). These results suggest that, rather than skewing the immune response, deletion of ATG16L1 compromises the ability of IECs to withstand damage. To test whether worsened disease is dependent on necroptosis signaling, we generated RIPK3-deficient Atg16L1ΔIEC mice (Atg16L1ΔIECRipk3−/−) and Cre-negative controls (Atg16L1f/fRipk3−/−) for comparison. Most Atg16L1ΔIECRipk3−/− mice survived allo-HCT and displayed a similar degree of disease as Atg16L1f/fRipk3−/− mice (Figure 1C-D). Additionally, Atg16L1ΔIEC mice treated with the RIPK1 inhibitor GSK547 exhibited significantly better survival and disease scores (Figure 1E-F). Thus, ATG16L1 protects against lethal GVHD by preventing RIPK1- and RIPK3-mediated necroptosis of IECs.
ATG16L1 prevents intestinal GVHD by inhibiting IEC necroptosis
We found that Atg16L1ΔIEC mice displayed shortening of the colon compared with controls, as well as exacerbated histopathology in the small intestine but not in the colon, liver, or skin (Figure 2A-B). ATG16L1 has a critical role in maintaining the viability and function of Paneth cells, secretory epithelial cells in the small intestinal crypts.13,16,27-30 Decreased Paneth cell numbers are observed in intestinal GVHD patients.31 Paneth cells are sensitive to endoplasmic reticulum stress because of their high secretory burden,32,33 and accumulation of damaged mitochondria upon autophagy inhibition contributes to loss of viability in a RIPK3-dependent manner.7,34 Consistent with this, Paneth cells, but not goblet cells, were significantly decreased in Atg16L1ΔIEC mice compared with controls (Figure 2C-D; supplemental Figure 2A-B). Paneth cell depletion in Atg16L1ΔIEC allo-HCT recipients was associated with an increase in terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)+ cells in the crypt base, whereas cleaved caspase-3 staining was minimal (Figure 2C-D), potentially reflecting nonapoptotic cell death.7,34-37 The colonic epithelium of Atg16L1ΔIEC mice also displayed an increase in TUNEL+ cells compared with Atg16L1f/f mice, but the total number of stained cells was modest and not as striking as in the small intestine (supplemental Figure 2C-D). RIPK3 deficiency reversed shortening of the colon, histopathology, Paneth cell depletion, and TUNEL staining in Atg16L1ΔIEC mice (Figure 2; supplemental Figure 2A-D). We also detected a RIPK3-dependent increase in bacteria in the spleen of Atg16L1ΔIEC mice following allo-HCT (supplemental Figure 2E). Collectively, these data indicate that inhibition of ATG16L1 in IECs exacerbates intestinal GVHD in an RIPK3-dependent manner.
![ATG16L1 prevents intestinal GVHD by inhibiting epithelial necroptosis. (A-D) Mice receiving BM and T cells from donor LP/J mice as in Figure 1 were euthanized on day 28 after allo-HCT and analyzed for signs of intestinal GVHD (n = 11 [Atg16L1f/f; f/f], n = 12 [Atg16L1ΔIEC; ΔIEC], n = 8 [f/f Ripk3−/−], and n = 8 [ΔIEC Ripk3−/−]). (A) Colon length. (B) Pathology score of small intestine, colon, liver, and skin. Representative images (C) and quantification (D) of hematoxylin and eosin (H&E), TUNEL, and cleaved caspase 3 staining. Arrowheads indicate Paneth cells or IECs positive for the indicated markers. Scale bars, 10 µm. At least 50 crypts were quantified per mouse. Data points in A, B, and D represent individual mice. Bars represent mean ± standard error of the mean, and ≥2 independent experiments were performed. *P < .05, **P < .01, ***P < .001, ****P < .0001. SI, small intestine.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/26/10.1182_blood.2019004116/1/m_bloodbld2019004116f2.png?Expires=1767745604&Signature=IErVBXDi6zQ-swZ0hao2PZRxbSrjaCGKQaU-LnkzTss5K20AVqPkQ~xukQiE67fQu~ZzYjLflaLLR1g2Aexcu9dF7XAUI57V1HVZzO50KQ~RiYCAv11FNLsULkCS46Q7eRHsiwLhezkj5Y0SHln3NzEPLjAnsbNhTV3vt3oCoDdwnZ9slWq5827s9n3r2m0WY6XOsQTLKsbK7PtX8LiyvzdBmcWQt-cpAvNiWKeW9b7GIhaCzYbuSsBZ09qnSQBgwI1ZKv6f4XWeqfZy0Mn-nMcsqle~7GU8LPu7feMGKvkRhgaUzY3A42jOTtkqiQeK6uZ~yUV6u8NDx-rwjLaLEw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ATG16L1 prevents intestinal GVHD by inhibiting epithelial necroptosis. (A-D) Mice receiving BM and T cells from donor LP/J mice as in Figure 1 were euthanized on day 28 after allo-HCT and analyzed for signs of intestinal GVHD (n = 11 [Atg16L1f/f; f/f], n = 12 [Atg16L1ΔIEC; ΔIEC], n = 8 [f/f Ripk3−/−], and n = 8 [ΔIEC Ripk3−/−]). (A) Colon length. (B) Pathology score of small intestine, colon, liver, and skin. Representative images (C) and quantification (D) of hematoxylin and eosin (H&E), TUNEL, and cleaved caspase 3 staining. Arrowheads indicate Paneth cells or IECs positive for the indicated markers. Scale bars, 10 µm. At least 50 crypts were quantified per mouse. Data points in A, B, and D represent individual mice. Bars represent mean ± standard error of the mean, and ≥2 independent experiments were performed. *P < .05, **P < .01, ***P < .001, ****P < .0001. SI, small intestine.
Allogeneic T cells injure intestinal organoids with autophagy gene mutations in vitro
Primary lymphocytes from mice that are added to intestinal organoid cultures retain viability and can differentiate.38,39 Whether such a coculture system can be used to assess lymphocyte effector functions and cytotoxicity is unknown. We established an ex vivo GVHD model by culturing organoids with T cells independently isolated from the spleen of allogeneic and syngeneic mice (supplemental Figure 3A). Small intestinal organoids derived from Atg16L1ΔIEC mice displayed a significant reduction in viability and surface area when cultured with allogeneic T cells (Figure 3A-C). By comparison, T cells had a minimal effect on Atg16L1f/f organoids. We confirmed the selective loss of viability displayed by Atg16L1ΔIEC organoids by measuring loss of metabolic activity through Thiazolyl Blue Tetrazolium Bromide (MTT) absorbance40 (supplemental Figure 3B-C). The susceptibility of Atg16L1ΔIEC organoids to cell death was dependent on alloreactivity, because syngeneic T cells from B6 mice did not significantly reduce viability or size (Figure 3A-C; supplemental Figure 3B-C). These data indicate that alloreactivity and genetic susceptibility can be recreated ex vivo.
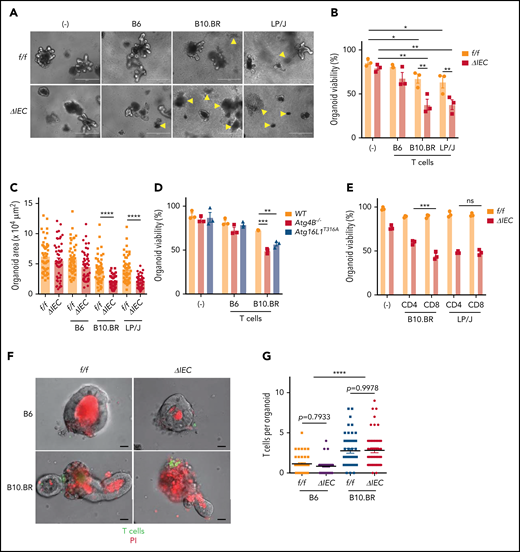
Allogeneic T cells induce cell death in intestinal organoids with autophagy gene mutations. Representative images (A), viability (B), and size (C) of small intestinal organoids from B6-background Atg16L1f/f (f/f) and Atg16L1ΔIEC (ΔIEC) mice cocultured for 48 hours with 1 × 105 splenic T cells separately harvested from B6, B10.BR, and LP/J mice. n = 3 mice each. Arrowheads indicate dead organoids. Scale bars, 400 µm. (D) Viability of organoids from B6-background Atg4B−/− and Atg16L1T316A mice cocultured for 48 hours with 1 × 105 splenic T cells separately harvested from B10.BR mice; n = 3 mice each. (E) Viability of small intestinal organoids from f/f and ΔIEC mice cocultured for 48 hours with FACS-sorted 1 × 105 CD4+ or 7 × 104 CD8+ T cells from B10.BR and LP/J mice; n = 3 mice each. Representative images (F) and number of T cells associated with organoid (G). At least 50 organoids were analyzed per group. T cells were stained with CellBrite Green (green) before coculture, and propidium iodide (PI; red) was added to the culture medium at the beginning to stain dead organoids/T cells. Scale bars, 25 µm; n = 3 mice each. Data points in B, D, and E are mean of technical replicates, and data points in C and F represent individual organoids. Bars represent mean ± standard error of the mean, and ≥2 independent experiments were performed. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant; WT, wild-type.
To confirm our findings, we examined the susceptibility of small intestinal organoids derived from Atg4B−/− mice, which lack another autophagy gene, and Atg16L1T316A mice, which harbor a knock-in mutation that mimics the human ATG16L1T300A variant. We found that Atg4B−/− and Atg16L1T316A B6 organoids displayed impaired viability compared with wild-type organoids when cultured with B10.BR T cells (Figure 3D). Both CD4+ and CD8+ T cells can contribute to GVHD, and depletion of CD4+ T cells (which also reduces the number of intestinal CD8+ T cells) was shown to prevent Paneth cell depletion.41 We found that CD8+ T cells sorted by flow cytometry (fluorescence-activated cell sorting) were more cytotoxic than CD4+ T cells when using B10.BR mice as the T-cell donor, whereas CD4+ and CD8+ T cells were equally capable of killing Atg16L1ΔIEC organoids when using LP/J mice as the T-cell donor; this suggests that the relative contribution of T-cell subpopulations may be donor strain specific (Figure 3E). T cells from B10.BR donors, but not syngeneic B6 donors, were physically associated with organoids when examined by light microscopy (Figure 3F-G). Autophagy proteins suppress MHC class I (MHC-I) levels in dendritic cells,42 thus raising the possibility that Atg16L1 deficiency also controls MHC-I in IECs. However, surface MHC-I was lower, rather than higher, in Atg16L1ΔIEC organoids compared with controls, and the number of T cells associated with both genotypes was similar (Figure 3F-G; supplemental Figure 3D), suggesting that genotype is not regulating the initial recognition of IECs by allogeneic T cells. Collectively, these data suggest that Atg16L1 deficiency causes organoids to become susceptible to the cytotoxic activity of allogeneic T cells.
Allogeneic T cells induce cytokine-mediated necroptosis in ATG16L1-deficient organoids
Atg16L1f/f and Atg16L1ΔIEC organoids containing allogeneic B10.BR or LP/J T cells had higher levels of TNF-α and IFN-γ compared with supernatant from those cultured with syngeneic B6 T cells or no T cells (Figure 4A). Organoids have been shown to be sensitive to these 2 cytokines.7,43,44 We also found that supernatant from organoids cocultured with B10.BR T cells contained higher levels of IL-22 (supplemental Figure 4A), which has been shown to exacerbate necroptosis in Atg16L1-mutant IECs.17 We tested the effect of blocking antibodies against TNF-α and IFN-γ, because these 2 cytokines were produced in the presence of B10.BR and LP/J T cells and, therefore, were most likely to mediate the effect of allogeneic T cells. Blocking TNF-α significantly increased survival of Atg16L1ΔIEC organoids, and blocking TNF-α and IFN-γ together completely rescued viability (Figure 4B). Additionally, we found that organoids from Atg16L1ΔIECRipk3−/− mice were resistant to B10.BR T-cell–mediated injury (Figure 4C-F; supplemental Figure 4B-C). These data are highly consistent with our in vivo results and support a model in which allogeneic T cells producing inflammatory cytokines induce necroptosis in ATG16L1-deficient IECs.
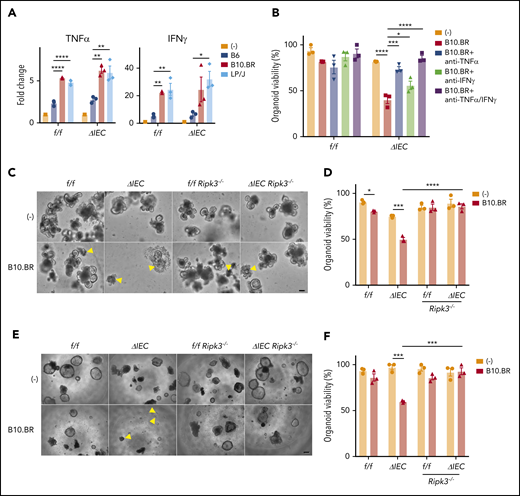
Allogeneic T cells induce TNF-α–mediated necroptosis in intestinal organoids. (A) Fold change in indicated cytokines in culture supernatants from Figure 3B. Each value is normalized to nonstimulated samples; n = 3 mice each. (B) Viability of small intestinal organoids treated or not with anti–TNF-α and/or anti–IFN-γ antibody and cocultured with B10.BR T cells for 48 hours; n = 3 mice each. Representative images of cocultured small intestinal (C) and colonic (E) organoids. Arrowheads denote dead organoids. Scale bars, 100 µm. Viability of small intestinal (D) and colonic (F) organoids from B6-background Atg16L1f/f (f/f), Atg16L1ΔIEC (ΔIEC), f/f Ripk3−/−, and ΔIEC Ripk3−/− mice cocultured for 48 hours with B10.BR T cells; n = 3 mice each. Data points in A, B, D, and F are mean of technical replicates. Bars represent mean ± standard error of the mean, and ≥2 independent experiments were performed. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Loss of viability in ATG16L1-deficient organoids is associated with an IFN signature
To examine the mechanism by which ATG16L1 deficiency renders IECs susceptible to necroptosis, we performed RNA sequencing analysis using Atg16L1f/f and Atg16L1ΔIEC small intestinal organoids, with or without TNF-α treatment. Principal component analysis shows that the samples cluster according to their condition, with cytokine stimulation and genotype statuses separating along PC1 and PC2, respectively (Figure 5A). In the absence of TNF-α, we found that 49 genes were upregulated at least twofold in Atg16L1ΔIEC organoids vs Atg16L1f/f organoids; many were known IFN-stimulated genes (ISGs) representing a type I IFN (IFN-I) signature (Figure 5B-C). ISGs remained upregulated in Atg16L1ΔIEC organoids treated with TNF-α (supplemental Figure 5A-B). We also found increased expression of genes associated with cytokine receptor signaling in TNF-α–treated organoids, but most of these were not impacted by Atg16L1 deficiency (supplemental Figure 5A-B; supplemental Table 2).
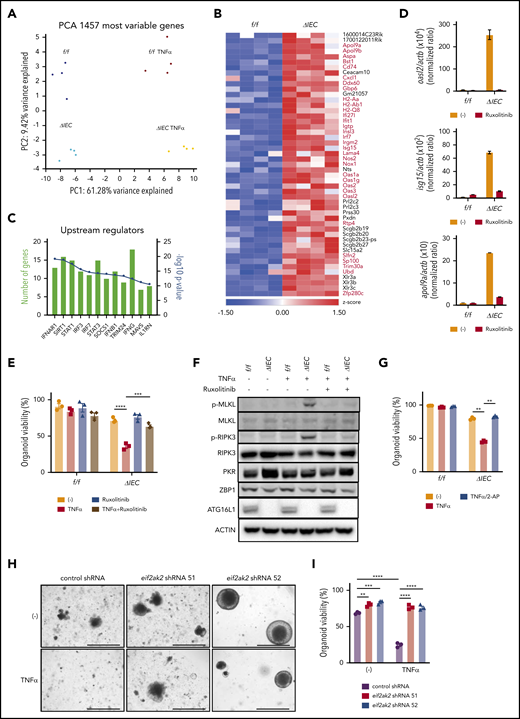
Loss of viability in ATG16L1-deficient intestinal organoids is associated with an IFN signature. (A) Unsupervised clustering based on expression of most variable genes by genotype and treatment with 20 ng/mL TNF-α for 2 hours. n = 4 replicates per group, each replicate was derived from separate mice. (B) Heat map of genes with a twofold change in Atg16L1ΔIEC (ΔIEC) over Atg16L1f/f (f/f) organoids. ISGs are highlighted with red and bold. (C) Pathway analysis of genes differentially expressed between f/f and ΔIEC naive organoids. (D) Quantitative reverse-transcription polymerase chain reaction (RT-PCR) measurement of indicated ISG expression normalized to actb in small intestinal organoids from B6 mice that were treated or not with 100 nM ruxolitinib at day 3. n = 3 mice each. (E) Viability of small intestinal organoids stimulated with 20 ng/mL TNF-α and/or 100 nM ruxolitinib for 48 hours. n = 3 mice each. (F) Western blot analysis of cell death–related proteins at day 3. f/f and ΔIEC organoids cultured with or without 100 nM ruxolitinib were treated with 20 ng/mL TNF-α for 2 hours. Blots are representative of ≥2 independent repeats. (G) Viability of small intestinal organoids stimulated with 20 ng/mL TNF-α and/or 500 μM 2-aminopurine (2-AP) for 48 hours. n = 3 mice each. Representative images (H) and viability (I) of small intestinal organoids from Atg16L1ΔIEC mice transduced with lentiviruses encoding shRNAs targeting Eif2ak2 or a nonspecific control and treated or not with 20 ng/mL TNF-α for 48 hours; n = 3 mice each. Scale bars, 1 mm. Data points in D, E, G, and I are mean of technical replicates. Bars represent mean ± standard error of the mean, and ≥2 independent experiments were performed. **P < .01, ***P < .001, ****P < .0001.
IFN-I activates JAK1 and STAT1/2 downstream of IFNAR1 to induce antiviral ISG expression, a pathway that has been shown to intersect necroptosis signaling.45-52 Cross talk between these signaling cascades promotes immunity during viral infection, potentially explaining why an antiviral cytokine contributes to an inflammatory form of programmed cell death.48,53,54 Although the role of IFN-I in GVHD is complex, because it can act on T cells or target tissue,55,56 recent studies reported the efficacy of JAK-STAT inhibitors, including ruxolitinib, in ameliorating GVHD in animal models and patients.57,58 We confirmed that Atg16L1ΔIEC organoids display an IFN-I signature by showing that 3 representative ISGs (Oasl2, Isg15, and Apol9a) are expressed at higher levels compared with controls, and we found that the expression of these genes can be inhibited by ruxolitinib (Figure 5D). Ruxolitinib also protected Atg16L1ΔIEC organoids from TNF-α–induced death and decreased phosphorylated MLKL and RIPK3 levels (Figure 5E-F). Next, we investigated whether a specific ISG might be involved in the increased susceptibility of Atg16L1ΔIEC organoids to necroptosis. Protein kinase R (PKR) encoded by Eif2ak2 is an ISG that was previously shown to license necroptosis downstream of JAK-STAT signaling and IFNs.59 We found that PKR (but not Z-DNA-binding protein 1 (ZBP1), another ISG implicated in necroptosis48,49,53 ) was increased in naive Atg16L1ΔIEC organoids, and the PKR-inhibitor 2-aminopurine (2-AP)59 completely protected Atg16L1ΔIEC organoids from TNF-α–induced death (Figure 5F-G). Atg16L1ΔIEC organoids transduced with 2 Eif2ak2 short hairpin RNAs (shRNAs), but not control shRNA, exhibited improved survival and resistance to TNF-α (Figure 5H-I; supplemental Figure 5C-D). Collectively, these results indicate that JAK-STAT signaling and PKR contribute to TNF-α–mediated necroptosis in ATG16L1-deficient organoids.
Development of an intestinal GVHD model using human intestinal organoids and peripheral T cells
Next, we examined whether human organoids generated from endoscopic biopsy specimens (supplemental Figure 6A; supplemental Table 3) display loss of viability when cultured with allogeneic T cells. Most biopsies were collected from Crohn’s disease patients because of their higher probability of harboring the ATG16L1T300A risk allele and the availability of small intestinal biopsies. Viable organoids can be generated from frozen tissue, allowing parallel experiments with banked immune cells60 or, in our case, coculture. T cells were sorted from PBMCs obtained from the same individuals as above or from an independent cohort of 20 healthy donors. To accurately compare viability in the presence of alloreactive T cells, it was necessary to culture all organoids in the presence of the same set of donor T cells. Therefore, PBMCs from the healthy donors were mixed prior to sorting T cells (supplemental Figure 6A). We confirmed that thawed organoids proliferated well in the absence of stimuli and verified the viability and purity of isolated T cells (supplemental Figure 6B-C). As an additional condition, we simultaneously evaluated the susceptibility of organoids to recombinant human TNF-α.
Allogeneic T cells were generally more toxic to human-derived organoids than to syngeneic ones (Figure 6A-B). Further, we found substantial variability in susceptibility to TNF-α or allogeneic T cells. Among the 20 small intestinal organoids that we tested, 15 exhibited a significant reduction in viability (75%) in the presence of allogeneic T cells, among which 6 displayed a high degree of susceptibility (30%), defined as a >50% loss in viability (Figure 6B-C; supplemental Figure 6D). Similarly, 15 displayed a significant reduction in viability (75%) and 7 displayed a high degree of susceptibility (46.7%) upon TNF-α treatment (Figure 6B-C; supplemental Figure 6D). Although organoids susceptible to allogeneic T cells were generally susceptible to TNF-α and vice versa, there were several examples for which individual organoids displayed noticeable differences in viability between these 2 treatments (patients 13, 19, and 20) (supplemental Figure 6E). Consistent with the observation that the small intestine was more susceptible to cell death and histopathology in Atg16L1ΔIEC allo-HCT recipient mice, we found that human colonic organoids were relatively resistant to killing by T cells or TNF-α (Figure 6D). Together, these findings establish a model to test IEC resilience to immune-mediated injury and show that organoids derived from humans display variability in susceptibility to killing by allogeneic T cells and TNF-α.
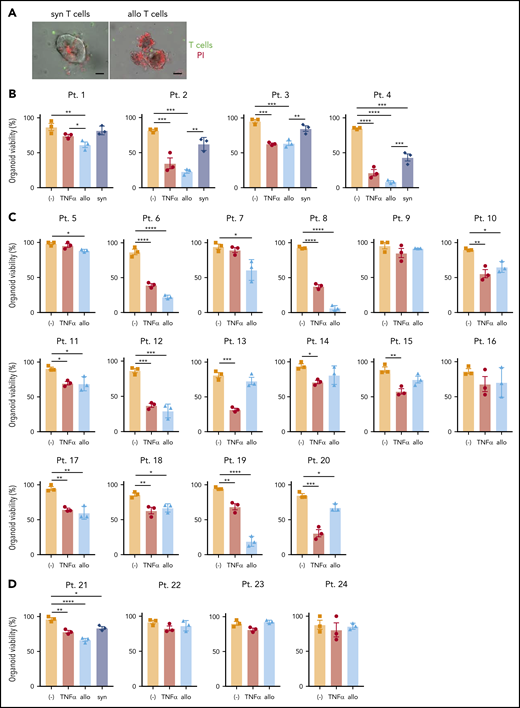
Development of an ex vivo intestinal GVHD model using human intestinal organoids and peripheral T cells. (A) Representative images of human small intestinal organoids cocultured for 8 hours with syngeneic (syn) or allogeneic (allo) human T cells. Sorted T cells were stained with CellBrite Green (green) before coculture, and PI (red) was added into the culture medium at the beginning to stain dead organoids. Scale bars, 25 µm. Viability of human small intestinal organoids from 20 patients (supplemental Table 3) at 48 hours after stimulation with 50 ng/mL TNF-α or post coculture with allogeneic and syngeneic T cells (B) or with only allogeneic T cells (C). (D) Viability of human colonic organoids from 4 patients (supplemental Table 2) at 48 hours after stimulation with 50 ng/mL TNF-α or post coculture with allogeneic and/or syngeneic T cells. At least 2 independent experiments were performed. *P < .05, **P < .01, ***P < .001, ****P < .0001. Pt., patient.
Intestinal organoids derived from ATG16L1T300A homozygous individuals display heightened susceptibility to death
Given that Atg16L1-mutant mouse organoids were susceptible to T-cell–mediated injury (Figure 3A-D), we hypothesized that the presence of the ATG16L1T300A risk allele contributes to the variability among human organoids. We retrospectively genotyped samples from the previous experiment for the presence of common single nucleotide polymorphisms (supplemental Table 3). Remarkably, almost all of the small intestinal organoids that displayed >50% death in the presence of TNF-α or allogeneic T cells were derived from individuals harboring 2 copies of the ATG16L1T300A risk variant (rs2241880), whereas most resistant organoids were from individuals with 0 or 1 copy of the allele (Figure 7A). Although several other IBD risk variants were present in our cohort, such as NOD2R702W (rs2066844), LRRK2N2081D (rs33995883), and IRGM (rs13361189), the organoids that were sensitive to cell death did not harbor these other risk variants (Figure 6B-C; supplemental Table 3). Analysis of our dataset, by comparing the degree of viability between individuals with 2 copies vs 0 or 1 copy of ATG16L1T300A, supported the conclusion that organoids from individuals who are homozygous for this allele display reduced survival in the presence of TNF-α or allogeneic T cells (Figure 7B).
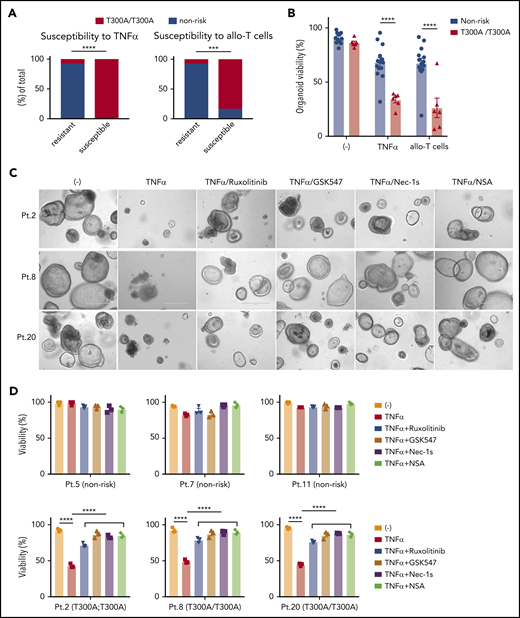
Intestinal organoids derived from ATG16L1T300Ahomozygous individuals exhibit heightened susceptibility to TNF-α and allogeneic T cells. (A) Proportion of human small intestinal organoids from Figure 6B and 6C that were susceptible (displayed >50% lethality) to recombinant TNF-α (left panel) or allogeneic T cells (right panel). n = 14 (nonrisk) and n = 6 (T300A/T300A). Statistical significance was validated with Fisher’s exact test. (B) Combined organoid viability in A; n = 14 (nonrisk) and n = 6 (T300A/T300A). Data points represent an average viability of individual organoids in Figure 6. Representative images (C) and viability (D) of human small intestinal organoids stimulated or not with 50 ng/mL TNF-α, 100 nM ruxolitinib, 1 μM GSK547, 20 μM necrostatin-1s (Nec-1s), or 2 μM necrosulfonamide (NSA) for 48 hours. Scale bars, 400 µm. Data points are mean of technical replicates. At least 2 independent experiments were performed. ***P < .001, ****P < .0001.
Finally, we examined whether drugs that target the underlying mechanism of susceptibility based on the mouse model would reverse the selective defect in viability displayed by ATG16L1T300A-homozygous human organoids. Specifically, we tested the efficacy of 2 RIPK1 inhibitors (necrostatin-1s and GSK547), an MLKL inhibitor (necrosulfonamide), and ruxolitinib. We used susceptibility to TNF-α, rather than allogeneic T cells, to avoid potential confounding effects of the drugs on T cells. At concentrations that are nontoxic to organoids from nonrisk patients, all 4 inhibitors significantly protected ATG16L1T300A-homozygous organoids from TNF-α–induced death (Figure 7C-D). These data indicate that ATG16L1 protects human IECs from TNF-α–mediated necroptosis, and necroptosis and JAK-STAT inhibitors could be promising therapeutic options for intestinal GVHD in patients with ATG16L1T300A risk alleles.
Discussion
Differential susceptibility of target tissues to injury potentially underlies heterogeneity in patients and represents an opportunity for designing individualized therapy. We showed that ATG16L1 has a conserved function in protecting IECs from killing by allogeneic T cells, such as those encountered following allo-HCT. ATG16L1 inhibited intestinal GVHD in a preclinical allo-HCT model by preventing necroptosis of IECs. Excess cell death in the intestinal epithelium resulting from dysregulated RIPK1 and RIPK3 signaling has been linked to intestinal inflammation,61-63 and inducing necroptosis by deleting caspase-8 in IECs is sufficient to induce a lethal inflammatory disease in mice along with Paneth cell depletion.64,65 We found that RIPK1/3 inhibition ameliorated GVHD and restored Paneth cells in allo-HCT recipient Atg16L1ΔIEC mice. Based on these results in the animal model, we were remarkably able to design an ex vivo GVHD platform that reproduced the role of ATG16L1 in IECs. Organoids from the small intestine of Atg16L1ΔIEC mice and ATG16L1T300A homozygous humans were susceptible to necroptosis induced by allogeneic T cells. Notably, we performed the experiments with human organoids blind to genotype rather than selecting for ATG16L1T300A homozygous samples. We believe that our findings provide proof-of-principle for a general approach in which heterogeneous responses to T-cell–mediated injury can be recreated in a quantitative ex vivo assay that can be used to identify variables that contribute to this interindividual variation.
Our results may have implications for the treatment of intestinal GVHD. When allogeneic T cells were cocultured with Atg16L1ΔIEC organoids, the combination of anti–TNF-α and anti–IFN-γ antibodies was required for full restoration of viability. Previous clinical studies revealed that TNF-α–targeted therapies, such as etanercept, a fusion protein of recombinant human soluble TNF-α, are promising,66-68 although still controversial,69 treatments for GVHD. Considering that the patients who participated in these studies were randomized and not separated by their ATG16L1 risk alleles, our results indicate that neutralization of TNF-α could be promising, especially for GVHD patients with ATG16L1T300A. Excess IL-22 has also been shown to induce necroptosis in ATG16L1-deficient IECs.17 It is possible that these and potentially other cytokines have redundant functions during intestinal GVHD and that blocking any individual cytokine would be insufficient to ameliorate disease. In this scenario, enhancing the resilience of the intestinal barrier to damage would be more efficacious. We also show that T cells can kill target cells through necroptosis. How ATG16L1 mutation facilitates this process is of great interest, because necroptosis frequently requires shunting of the pathway downstream of TNF-α away from apoptosis, such as through inhibition of caspase-8. ATG16L1 inhibition leads to STAT1 activation and ISG expression in IECs in a manner that is dependent on MAVS and STING, signaling adaptors involved in the sensing of viral nucleic acid.17,24 ATG16L1 and autophagy also mediate degradation of TRIF, an adaptor molecule involved in viral recognition,70 and Z-DNA-binding protein 1 (also known as DAI or DLM-1) interacts with RIPK3 to sensitize cells to virus-induced necroptosis.48,49,53 In this study, we found that Atg16L1ΔIEC organoids display increased levels of PKR, which is an RNA sensor and ISG, and inhibition of PKR significantly reduced necroptosis. Although multiple mechanisms downstream of IFN signaling likely contribute, our data suggest that PKR is 1 major IFN effector that is responsible for the increased susceptibility of ATG16L1-deficient organoids. More recently, IFN-λ, which also signals through STAT1, was shown to exacerbate necroptosis in IECs.71 Therefore, it is possible that inhibiting autophagy in IECs sensitizes cells to necroptosis by mimicking aspects of viral infection, such as activation of IFNs and JAK/STAT signaling.
Our results suggest that ATG16L1T300A-homozygous individuals are more likely to respond to therapies targeting RIPK1 or JAK/STAT signaling, both of which are in clinical trials for several diseases. Repurposing these drugs for treating intestinal GVHD or Crohn’s disease may be worth considering, especially if they can be targeted to likely responders. In summary, we suggest that advanced cell culture techniques that involve growing parenchymal cells, together with lymphocytes or their effector molecules, can recreate interindividual heterogeneity to tissue injury, which is a hallmark of a variety of disorders. This approach can be applied to multiple tissues. Parenchymal and lymphocyte specimens can be derived directly from the patient cohort of interest to predict susceptibility to injury for the purpose of prognosis or drug responsiveness.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the New York University Langone Health (NYULH) DART Microscopy Lab, NYULH Experimental Pathology Research Laboratory, NYU Langone’s Cytometry and Cell Sorting Laboratory, and NYU Langone's Genome Technology Center supported by the National Institutes of Health, National Cancer Institute grant P30 CA016087, and the Laboratory of Comparative Pathology and the Molecular Cytology Core and the Small-Animal Imaging Core Facilities supported by Memorial Sloan Kettering Cancer Center Support Grant/Core grant P30 CA008748) for advice and technical support. They also thank S. Fujii (Washington University School of Medicine, St. Louis, MO) for technical support in human organoid culture.
This work was supported in part by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL123340 (K.C., M.R.M.v.d.B.), R01 HL125816 (K.C.), R01 HL125571 (M.R.M.v.d.B.), and R01 HL147584 (M.R.M.v.d.B.); National Institute of Diabetes and Digestive and Kidney Diseases R01 DK093668 (K.C.) and R01 DK103788 (K.C.); National Institute of Allergy and Infectious Diseases R01 AI121244 (K.C., V.J.T.), R01 AI130945 (K.C., P.L.), U01 AI124275 (M.R.M.v.d.B); National Cancer Institute R01 CA228358 (M.R.M.v.d.B.), R01 CA228308 (M.R.M.v.d.B.), and P01 CA023766 (M.R.M.v.d.B.); and National Institute of Aging P01 AG052359 (M.R.M.v.d.B.). K.C. has also been supported by pilot awards from New York University Clinical and Translational Science Award UL1 TR001445 from the National Center for Advancing Translational Sciences and New York University Cancer Center Support Grant P30CA016087, National Institutes of Health; a Faculty Scholar grant from the Howard Hughes Medical Institute; the Kenneth Rainin Foundation; Institut Mérieux; and the Crohn’s & Colitis Foundation. M.R.M.v.d.B. has also been supported by The Lymphoma Foundation; The Susan and Peter Solomon Divisional Genomics Program; Tri-Institutional Stem Cell Initiative Award 2016-13; and the Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center. Y.M.-I. received a Research Fellowship Award from the Crohn’s & Colitis Foundation, and support grant from Japanese Society for the Promotion of Science (JSPS) Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers. K.C. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases, and has received support from the Stony-Wold Herbert Fund.
Authorship
Contribution: Y.M.-I., M.R.M.v.d.B., and K.C. formulated the original hypothesis, designed the study, analyzed the results, and wrote the manuscript; Y.M.-I. performed experiments and analyzed data; A.H. and P.L. established the clinical protocol and procured human material; Y.S. performed enzyme-linked immunosorbent assays and established the ex vivo platform; E.R. performed RNA sequencing analyses; A.L., J.J.T., and K.N. established the animal GVHD model; F.Y., J.A.N., X.Y., Y.-H.C., T.H., and S.L.S. analyzed the animal GVHD model; J.E.A. and D.H. harvested human endoscopic specimens; E.E.Z. and V.J.T. harvested PBMCs from healthy volunteers; M.Z.D. prepared histopathology samples; M.C. performed microscopic analyses; A.B., S.H., B.G., and J.B. performed experiments involving GSK547; C.L. analyzed histopathology samples; and all authors commented on the data and conclusions and approved the final version of the manuscript.
Conflict-of-interest disclosure: M.R.M.v.d.B. has consulted for, received honorarium or research support from, or participated in advisory boards for Seres Therapeutics, Flagship Ventures, Novartis, Evelo, Jazz Pharmaceuticals, Therakos, Amgen, Magenta Therapeutics, Merck & Co., Inc., Acute Leukemia Forum, PureTech, Straximm, Rubius Therapeutics, WindMIL Therapeutics, Mallinckrodt Pharmaceuticals, Kite Pharma Inc., and DKMS Medical Council (Board), and has intellectual property licensing agreements with Seres Therapeutics and Juno Therapeutics. K.C. has consulted for or received honoraria from Puretech Health, Genentech, and AbbVie and has a provisional patent, U.S. Patent Appln. No. 15/625 934. K.C. has received support from Pfizer Inc and AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Ken Cadwell, 430 East 29th St, 4th Floor, Laboratory 424, New York, NY 10016, e-mail: ken.cadwell@med.nyu.edu; and Marcel R.M. van den Brink, 1275 York Ave, New York, NY 10065, e-mail: vandenbm@mskcc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal