

In this issue of Blood, report and characterize novel and recurrent NTRK2/3 point mutations in ∼5% of patients with different hematologic neoplasms, including acute myeloid leukemia and lymphoblastic leukemia, as well as myeloproliferative disorders.1
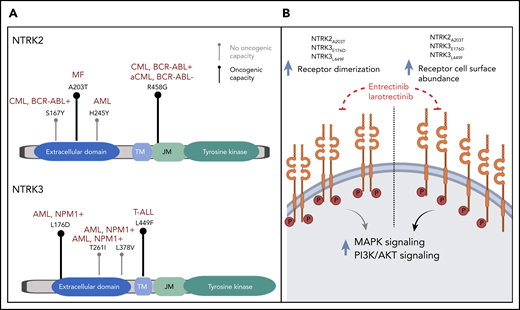
(A) Lollipop plots depicting the identified point mutations in NTRK2 (upper panel) and NTRK3 (lower panel) genes. Bold black marks indicate experimentally validated oncogenicity of the mutation; gray marks indicate failure to demonstrate oncogenicity. The diseases of patients in which the point mutations were identified are indicated in dark red. (B) Proposed mechanism by which the identified NTRK mutations increase oncogenicity. AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MF, myelofibrosis; T-ALL, T-cell acute lymphoblastic leukemia.
(A) Lollipop plots depicting the identified point mutations in NTRK2 (upper panel) and NTRK3 (lower panel) genes. Bold black marks indicate experimentally validated oncogenicity of the mutation; gray marks indicate failure to demonstrate oncogenicity. The diseases of patients in which the point mutations were identified are indicated in dark red. (B) Proposed mechanism by which the identified NTRK mutations increase oncogenicity. AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MF, myelofibrosis; T-ALL, T-cell acute lymphoblastic leukemia.
Molecular testing has evolved into an essential cornerstone in the diagnostic management of patients with both solid and hematologic malignancies. But how large should our testing panels be? Should we limit ourselves to target panels that include known driver genes that are associated with the disease? Should we include additional genes in cancer biology? Do we really need to analyze the entire coding sequence? There is always the hope of identifying unusual driver mutations that can be targeted with an inhibitor. But that hope stands against several variants of unknown significance that have no therapeutic consequence. Thus, logic and streamlined examples are needed to inform our approach to such data so that we can move from our large data collections toward actionable procedures that truly benefit our patients.
In their elegant experimental approach, Joshi et al demonstrated the oncogenicity of 4 of the 9 point mutations under consideration. They characterized the likely underlying patho-mechanism of those mutations, which led to increased cell surface abundance and receptor dimerization which, in turn, caused increased downstream pathway signaling (see figure). All 4 point mutations could successfully be treated with US Food and Drug Administration–approved NTRK inhibitors, and importantly, their antileukemic potential was effective even in the presence of mutations in other known driver oncogenes.
The identification and functional characterization as well as proof of targetability of the discovered NTRK point mutations is a highly important finding by itself. In contrast to solid tumors,2,3 the emphasis in hematologic malignancies has mainly been on NTRK fusion genes,4,5 which have been found to be responsive to NRTK inhibition.6-9 However, rare examples of somatic point variants warranted a study such as that by Joshi et al which had the goal of analyzing the frequency and relevance of NTRK point mutations.4,10 Given the observed frequency and especially the encouraging targetability of NTRK genes, adding those genes to targeted sequencing panels for hematologic malignancies should now be strongly considered.
The Joshi et al study has an impact that goes beyond that of the described NTRK genes, because the study reinforces general challenges and provides answers to recurring questions in the interpretation of variants detected in sequencing studies.
First, not all detected missense variants within a gene matter, and functional characterization is necessary to determine their relevance. Joshi et al found 9 different variants in 2 NTRK genes, but they showed that only 4 of the variants actually had oncogenic potential. Of note, some of these variants were located in highly conserved residues with predicted structure alterations via 3D modeling. This is not a novel concept, but it serves as a reminder that, even though validation experiments take time and effort, they are required in addition to commonly used in silico predictions.
Second, novel mutations that occur in the presence of a known driver mutation may substantially alter disease biology and may represent an important target lesion. In the patients with detected pathogenic NTRK mutations, at least 2 of them harbored BCR-ABL and CSF3R disease-defining mutations. This seems especially noteworthy for a disease like chronic myeloid leukemia; in the relapse setting, the focus is on detecting known resistance mutations and does not routinely include a broader screening approach.
So large-scale genomic profiling is indeed tedious, often yields results only in mutations of known foes, and is very expensive. But Joshi et al provide a very good example of why, if done the right way, this approach is necessary not only for personalized patient care but also to enhance our understanding of disease biology.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal