

Key Points
Antiviral responses impact the efficacy of immunoprophylaxis against the KEL glycoprotein antigen on RBCs in mice.
The immunoprophylaxis failure induced by poly(I:C) or type 1 IFN occurs even in the absence of recipient type 1 IFN receptors.

Abstract
Polyclonal anti-D (Rh immune globulin [RhIg]) therapy has mitigated hemolytic disease of the newborn over the past half century, although breakthrough anti-D alloimmunization still occurs in some treated females. We hypothesized that antiviral responses may impact the efficacy of immunoprophylaxis therapy in a type 1 interferon (IFN)-dependent manner and tested this hypothesis in a murine model of KEL alloimmunization. Polyclonal anti-KEL immunoprophylaxis (KELIg) was administered to wild-type or knockout mice in the presence or absence of polyinosinic-polycytidilic acid (poly[I:C]), followed by the transfusion of murine red blood cells (RBCs) expressing the human KEL glycoprotein. Anti-KEL alloimmunization, serum cytokines, and consumption of the transfused RBCs were evaluated longitudinally. In some experiments, recipients were treated with type 1 IFN (IFN-α/β). Recipient treatment with poly(I:C) led to breakthrough anti-KEL alloimmunization despite KELIg administration. Recipient CD4+ T cells were not required for immunoprophylaxis efficacy at baseline, and modulation of the KEL glycoprotein antigen occurred to the same extent in the presence or absence of recipient inflammation. Under conditions where breakthrough anti-KEL alloimmunization occurred, KEL RBC consumption by inflammatory monocytes and serum monocyte chemoattractant protein-1 and interleukin-6 were significantly increased. Poly(I:C) or type I IFN administration was sufficient to cause breakthrough alloimmunization, with poly(I:C) inducing alloimmunization even in the absence of recipient type I IFN receptors. A better understanding of how recipient antiviral responses lead to breakthrough alloimmunization despite immunoprophylaxis may have translational relevance to instances of RhIg failure that occur in humans.
Introduction
Red blood cell (RBC) alloimmunization, which occurs following exposure to non-self RBC antigens in transfusion or pregnancy, can be clinically significant in both settings. Pregnancy is a relatively common source of RBC alloimmunization in females,1 given its prevalence in comparison with transfusion. Rh immune globulin (RhIg) is the only therapy known to prevent alloimmunization during pregnancy in humans and is one of the most successful antibody-based therapies developed to date, significantly decreasing anti-D–mediated hemolytic disease of the fetus and newborn. Although widely used for half of a century,2 the mechanism of action of RhIg remains poorly understood.3,4
We have previously described a murine model of immunoprophylaxis, developed as a surrogate to investigate the potential mechanism(s) of action of RhIg. In this model, polyclonal antibodies against the KEL glycoprotein (KELIg) prevent the development of anti-KEL alloantibodies.5 The mechanism of action of KELIg remains unclear, with 1 possibility being that the recipient’s immune system does not fully recognize the KEL RBC antigen, as KELIg causes the KEL antigen on circulating RBCs to be altered to the point of being undetectable within hours of transfusion (eg, antigen modulation).6 Antibodies against other RBC antigens in murine models have also been found to suppress immune responses,7,8 although the mechanism(s) have not been fully elucidated.
Despite the tremendous success of RhIg immunoprophylaxis in humans, breakthrough anti-D alloimmunization still occurs.9 Furthermore, as RhIg is generated in RhD-negative human volunteers, the supply remains inadequate to meet the worldwide need. A better understanding of how antibody-mediated immunoprophylaxis prevents RBC alloimmunization may not only mitigate failures but also inform implementation of alternate therapies, including the use of more readily available recombinant antibodies.
The importance of the recipient inflammatory state on RBC alloimmunization has previously been documented in murine models and in humans.10-12 Recent studies have identified the importance of type 1 interferon (IFN-α/β) production and signaling through the IFN-α receptor (IFNAR1) on RBC alloimmunization.10,13,14 Furthermore, acute viral infection is common in humans and associated with increased levels of IFN-α/β and IFN-stimulated genes.15 In this paper, we hypothesized that recipient antiviral responses cause immunoprophylaxis to fail, in part, by an IFN-α/β–dependent mechanism. Polyinosinic-polycytidilic acid (poly[I:C]), an analog of double-stranded RNA, was used as a viral mimic in this murine model, as it has been shown to enhance alloimmune responses in other settings.10,16 The results of these studies may have translational relevance to RhIg successes and failures.
Methods
Mice
C57BL/6 mice were purchased from Charles River (Wilmington, MA), and T-cell receptor (TCR) α−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Ifnar1−/−, Irf3−/−, and Irf7−/− mice were previously described.17-19 Appropriate gene-deficient mice were bred to produce Irf3/7−/− double knockout mice. Animals lacking Irf3/Irf5/Irf7 were generated by Michael Diamond and generously provided by Sujan Shresta. Transgenic mice expressing the entire human KEL glycoprotein were generated and bred by our laboratory. The mice used for these experiments have been previously described as “KEL2B” and express the KEL2 antigen in addition to the Jsb antigen, the Kpb antigen, and other antigens in the KEL family on their RBCs.20 In this study, they are referred to as “KEL” mice as the protein being studied includes the entire human KEL glycoprotein. All animals were housed in Yale University’s animal facilities. All mice were 8 to 12 weeks of age and were backcrossed to the C57BL/6 background for >8 generations. All procedures and protocols were approved by Yale University’s Institutional Care and Use Committee.
KELIg generation and immunization
Polyclonal KELIg antisera was generated as previously described,5 by transfusing transgenic KEL RBCs into C57BL/6 recipients pretreated with an intraperitoneal injection of 100 μg of high-molecular-weight poly(I:C) (Invivogen, San Diego, CA) a total of 3 times, separated by 2 weeks between each transfusion. Pooled sera collected 2 to 4 weeks after the final transfusion was tested for KEL binding ability by flow crossmatch with KEL or control C57BL/6 RBCs as targets; all immunoglobulin G (IgG) subtypes are represented.5 Following dose titration experiments, recipient mice were passively transferred with enough KELIg to lead to maximal RBC clearance (between 10 and 20 µL per experiment). In some experiments, recipient CD4+ T cells were depleted by injection with GK1.5 (BioXcell), as previously described.21,22
Blood collection, labeling, and transfusion
Donor KEL or wild-type C57BL/6 RBCs were collected into anticoagulant preservative solution (citrate phosphorus dextrose adenine; Jorgensen Labs, Henry Schein, Melville, NY), leukoreduced over a syringe filter (Pall Corporation, Port Washington, NY), and washed to remove residual citrate. Prior to transfusion in some experiments, RBCs were labeled with chloromethylbenzamido 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) or 3,3′-dihexadecyloxacarbocyanine perchlorate (DiO) according to the manufacturer’s instructions (Molecular Probes, Eugene, OR) and as previously described.23 In other experiments, 100 × 103 units of recombinant mouse IFN-α (HC1040; Hycult Biotech, Uden, The Netherlands) was cotransfused. Recipient mice were transfused via lateral tail vein with an equivalent of 1 unit of KEL RBCs (as well as control RBCs in RBC recovery experiments). Survival of the transfused RBCs was determined by calculating the ratio of circulating DiO to DiI RBCs in recipients at select time points posttransfusion.
Flow cytometry
To evaluate the active immune response to mice after transfusion, sera was collected at multiple time points, and the anti-KEL IgG responses were measured using a flow cytometric crossmatch assay with antigen-positive (KEL) or antigen-negative (C57BL/6) RBC targets. Secondary antibodies included goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA). The antigen-specific response (adjusted mean fluorescence intensity) was determined by subtracting the signal of serum with antigen-negative C57BL/6 RBCs from that of serum with antigen-positive RBCs. Transfused RBCs were analyzed for the KEL antigen by incubating them with KELIg followed by anti-mouse IgG, or a recombinant anti-KEL (anti-CD238, clone REA330; Miltenyi, Sommerville, MA) or the isotype matched control.
Following transfusion of DiO-labeled RBCs, splenic cell subsets were evaluated. Spleens were harvested into ice-cooled RPMI 1640 media and finely minced with a razor blade. Then, single-cell suspensions of splenocytes were prepared by passing the samples through a 70-µm nylon cell strainer and collecting them on ice-cold fluorescence-activated cell sorter buffer (Dulbecco's phosphate-buffered saline modified, 0.2% bovine serum albumin, 0.5 M EDTA). The RBCs were lysed with ammonium chloride, and splenocytes were treated with Fc block (anti-mouse CD16/CD32; BD Biosciences, San Jose, CA) followed by incubation with cell-surface antibodies. Antibodies against CD19 (clone 6D5), TCRβ (clone H57-597), CD11b (clone M1/70), CD11c (clone N418), CD8a (clone 53-6.7), F4/80 (clone BM8), CD115 (CSF-1R, clone AFS98), Ly-6G/Ly-6C (GR-1, clone RB6-8C5), and PDCA-1 (CD137, BST2, clone 927) were purchased from BioLegend (San Diego, CA). Anti-TER119 (clone TER119) was purchased from Invitrogen (Thermo Fisher Scientific, Carlsbad, CA). Zombie Violet Viability Dye was purchased from BioLegend. Spleen samples were analyzed on a BD LSR II cytometer; other samples were analyzed on a BD FACSCalibur or a Miltenyi MACSQuant.
Cytokine/chemokine measurements by bead based immunoassay
Serum cytokines and chemokines were measured using the multiplex immunoassay LEGENDplex Mouse Inflammation Panel (BioLegend) following the manufacturer’s instructions. Samples were read in a BD FACSCalibur. Data analysis was performed with the LEGENDplex Data Analysis Software v.8 (BioLegend).
Statistics
All statistical analysis was performed using Graph Pad Prism software (San Diego, CA). Statistical significance between 2 groups of nonparametric data was determined using a Mann-Whitney U test, and statistical significance between 3 or more groups was determined using the Kruskal-Wallis test with Dunn's posttest. Error bars represent 1 standard deviation, and significance was determined by a value of P < .05. The bars on each graph indicate means, and the dots indicate data from individual mice.
Results
KELIg immunoprophylaxis prevents anti-KEL alloimmunization independently of recipient CD4+ T cells
We have previously described the efficacy of passively administered polyclonal anti-KEL (KELIg) at preventing alloimmunization to transfused KEL RBCs in wild-type recipients.5,6 As a general experimental design, KELIg was passively administered to the recipients by tail vein injection on day −1. On day 0, recipient mice were transfused with leukoreduced KEL RBCs. The transfusion recipient’s anti-KEL response was evaluated longitudinally thereafter (Figure 1A).
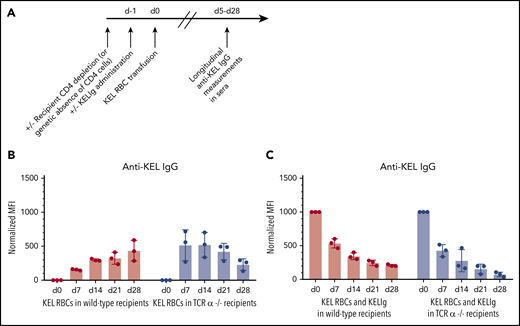
KELIg prevents alloimmunization independently of recipient CD4+ T cells. (A) General experimental design: recipients depleted of CD4+ T cells were transfused with KEL RBCs in the presence or absence of KELIg, and anti-KEL IgG alloimmune responses were measured longitudinally in serum posttransfusion. (B) Total anti-KEL IgG measured in the serum of recipients following a KEL RBC transfusion in the absence of KELIg, or (C) in the presence of KELIg. These data are representative of 3 independent experiments with 3 to 5 mice per group per experiment (in total, 11 and 11 mice were studied across 3 experiments in panel B with the same number studied in panel C). There were no statistically significant differences between groups; error bars indicate standard deviation between individual mice. MFI, mean fluorescence intensity.
KELIg prevents alloimmunization independently of recipient CD4+ T cells. (A) General experimental design: recipients depleted of CD4+ T cells were transfused with KEL RBCs in the presence or absence of KELIg, and anti-KEL IgG alloimmune responses were measured longitudinally in serum posttransfusion. (B) Total anti-KEL IgG measured in the serum of recipients following a KEL RBC transfusion in the absence of KELIg, or (C) in the presence of KELIg. These data are representative of 3 independent experiments with 3 to 5 mice per group per experiment (in total, 11 and 11 mice were studied across 3 experiments in panel B with the same number studied in panel C). There were no statistically significant differences between groups; error bars indicate standard deviation between individual mice. MFI, mean fluorescence intensity.
First, baseline responses to transfused KEL RBCs were established in the absence of KELIg. Figure 1B shows the general kinetics of anti-KEL alloantibody responses following a KEL RBC transfusion in wild-type recipients or in animals genetically lacking conventional CD4+ T cells expressing the αβ receptor (TCRα−/−). These data are similar to those that we have previously observed,24 with 100% of wild-type recipients generating anti-KEL IgG alloantibodies and with this primary antibody response occurring despite a lack of recipient CD4+ T cells.25,26
To investigate whether recipient CD4+ T cells were required for immunoprophylaxis efficacy, animals genetically lacking CD4+ T cells or wild-type animals depleted of CD4+ T cells were treated with KELIg and transfused with KEL RBCs. Regardless of the approach taken to eliminate recipient CD4+ T cells (Figure 1C shows TCRα−/− data), KELIg effectively prevented alloimmunization. As supported by prior studies,5 the day 0 time point in Figure 1C and other figures is a measure of passive anti-KEL detected following KELIg administration; the day 14 time point represents a mixture of passively administered KELIg and actively formed anti-KEL, and the day 28 time point represents primarily actively formed anti-KEL.
Poly(I:C) causes breakthrough anti-KEL alloimmunization despite KELIg immunoprophylaxis
We and others have observed that recipient inflammation enhances RBC alloantibody responses.10,16 As such, we hypothesized that recipient treatment with poly(I:C), a synthetic compound mimicking viral RNA and known to induce an antiviral IFN response, may cause KELIg immunoprophylaxis to fail. Using the experimental design shown in Figure 2A, KELIg was administered to wild-type animals, which were then treated or not treated with a single dose of poly(I:C) prior to KEL RBC transfusion. Whereas treatment with KELIg immunoprophylaxis prevented active anti-KEL alloimmunization in untreated recipients, breakthrough anti-KEL alloimmunization occurred in recipients treated with poly(I:C) 2 to 4 hours prior to the KEL RBC exposure (Figure 2B).

Poly(I:C) causes breakthrough anti-KEL alloimmunization despite KELIg immunoprophylaxis. (A) General experimental design: recipients were passively immunized with KELIg, followed by treatment with poly(I:C) or a saline control, followed by transfusion with murine RBCs expressing the human KEL glycoprotein. Alloimmune responses were measured longitudinally in serum posttransfusion. (B) Total anti-KEL IgG measured in the serum of recipients from day 0 to day 28 posttransfusion. (C) Experiment altering the timing of poly(I:C) administration; in this instance, poly(I:C) was administered 4 hours before or 36 hours following KEL RBC transfusion. (D) Animals depleted of CD4+ T cells using GK1.5 antibody were treated with KELIg and transfused in the presence or absence of poly(I:C), with anti-KEL IgG measured longitudinally. These data are representative of 2 to 3 independent experiments, with 3 to 6 mice per group per experiment (in total, 13 and 15 mice were studied across 3 experiments for panel A; 6 and 6 mice were studied across 2 experiments for panel B; 11 and 10 mice were studied across 3 experiments for panel C). *P < .05; error bars indicate standard deviation between mice.
Poly(I:C) causes breakthrough anti-KEL alloimmunization despite KELIg immunoprophylaxis. (A) General experimental design: recipients were passively immunized with KELIg, followed by treatment with poly(I:C) or a saline control, followed by transfusion with murine RBCs expressing the human KEL glycoprotein. Alloimmune responses were measured longitudinally in serum posttransfusion. (B) Total anti-KEL IgG measured in the serum of recipients from day 0 to day 28 posttransfusion. (C) Experiment altering the timing of poly(I:C) administration; in this instance, poly(I:C) was administered 4 hours before or 36 hours following KEL RBC transfusion. (D) Animals depleted of CD4+ T cells using GK1.5 antibody were treated with KELIg and transfused in the presence or absence of poly(I:C), with anti-KEL IgG measured longitudinally. These data are representative of 2 to 3 independent experiments, with 3 to 6 mice per group per experiment (in total, 13 and 15 mice were studied across 3 experiments for panel A; 6 and 6 mice were studied across 2 experiments for panel B; 11 and 10 mice were studied across 3 experiments for panel C). *P < .05; error bars indicate standard deviation between mice.
To determine if the timing of poly(I:C) administration in relationship to RBC exposure10 is critical in the immunoprophylaxis model, poly(I:C) was given 2 to 4 hours prior to KEL RBC exposure or 36 hours after KEL RBC exposure. Unlike what was observed in the animals receiving poly(I:C) in close proximity to the transfused RBCs, breakthrough alloimmunization did not occur in the group receiving poly(I:C) 36 hours after the transfused RBCs (Figure 2C).
Given prior results demonstrating that CD4+ T cells are required for the ability of poly(I:C) to enhance KEL RBC-induced antibody formation in the absence of KELIg (Patel et al27 ; supplemental Figure 1, available on the Blood Web site), we investigated whether recipient CD4+ T cells play a role in the breakthrough alloimmunization observed in the presence of poly(I:C) in the studied model. Animals lacking CD4+ T cells were treated with KELIg in the presence or absence of poly(I:C), with alloimmune responses evaluated longitudinally. There were no differences in anti-KEL responses between these 2 groups (Figure 2D). These data, taken together with the data shown in Figure 2B, suggest that CD4+ T cells promote poly(I:C)-breakthrough alloimmunization even in the presence of KELIg immunoprophylaxis.
KEL RBC clearance rates and antigen modulation are similar in the presence or absence of poly(I:C) and KELIg immunoprophylaxis
Clearance of antibody-coated incompatible RBCs has been hypothesized as a mechanism of immunoprophylaxis efficacy, although the importance of RBC clearance remains unclear in animal models and in humans alike.28 We have previously shown that KELIg leads to the rapid clearance of ∼50% of KEL-expressing RBCs, whereas the transfused cells that remain in circulation no longer express detectable KEL glycoprotein.5 Given the breakthrough alloimmunization observed in the presence of poly(I:C), we hypothesized that poly(I:C) impacted clearance patterns or antigen expression on the RBCs remaining in circulation. To evaluate clearance, KEL RBCs were labeled with the lipophilic dye DiO and mixed with wild-type RBCs labeled with the lipophilic dye DiI. Following treatment and transfusion as per the schematic in Figure 2A, RBCs were recovered from wild-type recipients at 10 minutes, 1 hour, and 24 hours posttransfusion, and the ratio of DiO KEL RBCs to DiI wild-type RBCs was evaluated. KEL RBCs cleared rapidly in recipients treated with KELIg immunoprophylaxis, with no differences observed in the presence or absence of recipient poly(I:C) treatment (Figure 3A).

KEL RBC clearance and modulation of the KEL antigen in the setting of KELIg immunoprophylaxis are similar with or without poly(I:C). (A) KEL RBCs labeled with the lipophilic dye DiO were mixed with wild-type RBCs labeled with the lipophilic dye DiI and transfused into recipients treated with KELIg, in the presence or absence of KELIg; posttransfusion RBC recovery and survival were measured and are presented as a ratio of KEL RBCs to wild-type RBCs. (B) Recovered DiO-labeled KEL RBCs were evaluated for KEL glycoprotein antigen expression after incubation with KELIg and a fluorescently conjugated anti-mouse IgG. (C) Recovered DiO-labeled KEL RBCs in animals treated with or without KELIg were also evaluated for KEL glycoprotein expression using a recombinant antibody against CD238. The data in panels A and B are representative of 3 independent experiments with 3 mice per group per experiment (total of 9 and 9 mice); the data in panel C are representative of 2 independent experiments with 2 to 3 mice per group per experiment (total of 5 and 5 mice); error bars indicate standard deviation between mice.
KEL RBC clearance and modulation of the KEL antigen in the setting of KELIg immunoprophylaxis are similar with or without poly(I:C). (A) KEL RBCs labeled with the lipophilic dye DiO were mixed with wild-type RBCs labeled with the lipophilic dye DiI and transfused into recipients treated with KELIg, in the presence or absence of KELIg; posttransfusion RBC recovery and survival were measured and are presented as a ratio of KEL RBCs to wild-type RBCs. (B) Recovered DiO-labeled KEL RBCs were evaluated for KEL glycoprotein antigen expression after incubation with KELIg and a fluorescently conjugated anti-mouse IgG. (C) Recovered DiO-labeled KEL RBCs in animals treated with or without KELIg were also evaluated for KEL glycoprotein expression using a recombinant antibody against CD238. The data in panels A and B are representative of 3 independent experiments with 3 mice per group per experiment (total of 9 and 9 mice); the data in panel C are representative of 2 independent experiments with 2 to 3 mice per group per experiment (total of 5 and 5 mice); error bars indicate standard deviation between mice.
The recovered DiO-labeled KEL RBCs were also evaluated for the presence of the KEL glycoprotein antigen, by incubating the recovered cells with anti-KEL antibody followed by fluorescently labeled mouse IgG. As previously described, antigen expression remained stable at 10 minutes, 1 hour, and 24 hours posttransfusion in animals transfused in the absence of KELIg5 (data not shown). In contrast, the antigen rapidly became undetectable in animals treated with KELIg. There were no detectable differences in antigen expression in recipients treated with KELIg in the presence or absence of poly(I:C) (Figure 3B). The lack of detectable KEL antigen was confirmed using a different anti-KEL detection reagent, a recombinant antibody against the KEL glycoprotein (Figure 3C). Taken together, these data show no consistent patterns between RBC clearance, antigen detection, and immunoprophylaxis efficacy.
Poly(I:C) and KELIg immunoprophylaxis shift phagocytosis of transfused KEL RBCs away from CD8α DCs and toward inflammatory monocytes
Despite similar peripheral blood RBC clearance patterns, we hypothesized that transfused RBCs would be shunted to different phagocytic cell subsets under different conditions. To test this hypothesis, experiments were set up as per Figure 2A, using DiO-labeled KEL RBCs. At early (3 hours) and later (16 hours) time points posttransfusion, splenic cell subsets were quantitated and evaluated for DiO fluorescence (indicating consumption of KEL RBCs). After gating on single cells, live cells, and non-T cells/non-B cells/non-RBCs, we evaluated splenic red pulp macrophages, dendritic cells ([DCs]: CD8α DCs, CD11b DCs, and plasmacytoid DCs), resident and inflammatory monocytes, and neutrophils (Figure 4A). There were changes in the general composition of splenic cell subsets recovered from the spleens of animals treated with KELIg and poly(I:C) compared with KELIg alone (Figure 4B-C), similar to those we have previously described after poly(I:C) treatment alone.29 More pronounced, however, were the changes in the detection of DiO-labeled KEL RBCs inside these cell subsets. In the setting of KELIg without poly(I:C), for example, CD8α DCs along with the other shown populations had significantly more DiO signal compared with the setting of KELIg with poly(I:C), 3 hours after transfusion (Figure 4E). By 16 hours after transfusion, red pulp macrophages, inflammatory monocytes, and neutrophils (among other cell subsets) had significantly more DiO signal in the setting of KELIg with poly(I:C) compared with the setting of KELIg without poly(I:C) (Figure 4D,F). These findings are consistent with these splenic cell subsets consuming a greater number of DiO-labeled KEL RBCs, having slower kinetics of the destruction of consumed DiO-labeled KEL RBCs, or a combination of the two.

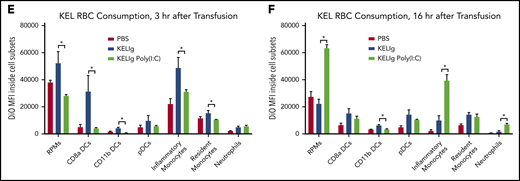
Poly(I:C) and KELIg immunoprophylaxis shift phagocytosis of transfused KEL RBCs away from CD8α DCs and toward inflammatory monocytes. (A) Gating strategy for splenic cell subsets. DiO-labeled KEL RBCs were transfused to animals treated with KELIg in the presence or absence of poly(I:C), and splenic cell subsets were evaluated at 3 and 16 hours posttransfusion (B-C). (D) Representative histograms for DiO RBC fluorescence patterns of splenic cell subsets, after first excluding TER119-positive RBCs on the exterior of the splenic cells, and DiO expression by the splenic cell subsets evaluated at 3 and 16 hours posttransfusion (E-F). These data are representative of 2 to 3 independent experiments with 3 mice per group per experiment (in total, 9, 9, and 9 mice were studied across 3 experiments for panels B and E; 6, 6, and 6 mice were studied across 2 experiments for panels C and F). *P < .05 between KELIg and KEL RBCs in the presence or absence of poly(I:C); error bars indicate standard deviation between mice. FSC, forward scatter; PBS, phosphate-buffered saline; pDCs, plasmacytoid dendritic cells; RPMs, red pulp macrophages.
Poly(I:C) and KELIg immunoprophylaxis shift phagocytosis of transfused KEL RBCs away from CD8α DCs and toward inflammatory monocytes. (A) Gating strategy for splenic cell subsets. DiO-labeled KEL RBCs were transfused to animals treated with KELIg in the presence or absence of poly(I:C), and splenic cell subsets were evaluated at 3 and 16 hours posttransfusion (B-C). (D) Representative histograms for DiO RBC fluorescence patterns of splenic cell subsets, after first excluding TER119-positive RBCs on the exterior of the splenic cells, and DiO expression by the splenic cell subsets evaluated at 3 and 16 hours posttransfusion (E-F). These data are representative of 2 to 3 independent experiments with 3 mice per group per experiment (in total, 9, 9, and 9 mice were studied across 3 experiments for panels B and E; 6, 6, and 6 mice were studied across 2 experiments for panels C and F). *P < .05 between KELIg and KEL RBCs in the presence or absence of poly(I:C); error bars indicate standard deviation between mice. FSC, forward scatter; PBS, phosphate-buffered saline; pDCs, plasmacytoid dendritic cells; RPMs, red pulp macrophages.
Multiple inflammatory cytokines are induced by poly(I:C) in the setting of KELIg immunoprophylaxis
Cytokines present in humans after treatment with RhIg have previously been evaluated,30 and the inflammatory cytokines induced by poly(I:C) are also well described.31 However, the recipient cytokines induced by KELIg or by the combination of KELIg and poly(I:C) have not been reported. Thus, we evaluated serum cytokines at early (0.5 hours and 1 hour) and later (16 to 24 hours) time points after KEL RBC transfusion alone or after KELIg and KEL RBC transfusion in the presence or absence of poly(I:C). Transfusion alone did not significantly alter the tested serum cytokines above baseline (Figure 5, group labeled PBS). KELIg and KEL RBCs led to moderate early increases in IFN-γ and tumor necrosis factor-α (TNF-α), followed by detectable interleukin (IL-6) and monocyte chemoattractant protein-1 (MCP-1) at later time points (Figure 5, group labeled KELIg). In the presence of poly(I:C), however, there was a more marked increase in IL-6 and MCP-1 (Figure 5, group labeled KELIg Poly(I:C)).
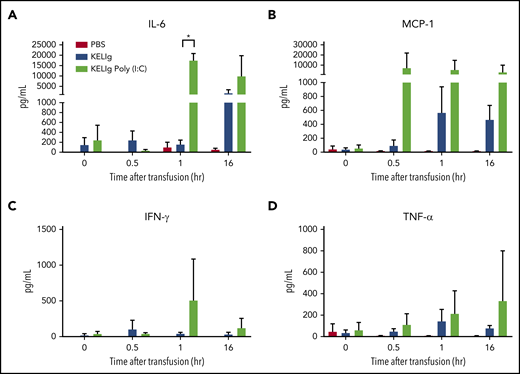
Multiple inflammatory cytokines are induced by poly(I:C) in the setting of KELIg immunoprophylaxis. Animals were treated with KELIg immunoprophylaxis and transfused with KEL RBCs, in the presence or absence of poly(I:C). Serum cytokines were evaluated prior to transfusion and at 0.5, 1, and 16 hours after transfusion using the LEGENDplex kit. (A) IL-6, (B) MCP-1, (C) IFN-γ, and (D) TNF-α are shown. There were no differences observed in the 2 other cytokines (IL-10 and IL-12) evaluated. These data are representative of 2 independent experiments with 3 mice per group per experiment using LEGENDplex and 2 additional experiments with 3 mice per group per experiment using a different platform (in total, 12, 12, and 12 mice were studied across 4 experiments). *P < .05 between KELIg and KEL RBCs in the presence or absence of poly(I:C); error bars indicate standard deviation between mice.
Multiple inflammatory cytokines are induced by poly(I:C) in the setting of KELIg immunoprophylaxis. Animals were treated with KELIg immunoprophylaxis and transfused with KEL RBCs, in the presence or absence of poly(I:C). Serum cytokines were evaluated prior to transfusion and at 0.5, 1, and 16 hours after transfusion using the LEGENDplex kit. (A) IL-6, (B) MCP-1, (C) IFN-γ, and (D) TNF-α are shown. There were no differences observed in the 2 other cytokines (IL-10 and IL-12) evaluated. These data are representative of 2 independent experiments with 3 mice per group per experiment using LEGENDplex and 2 additional experiments with 3 mice per group per experiment using a different platform (in total, 12, 12, and 12 mice were studied across 4 experiments). *P < .05 between KELIg and KEL RBCs in the presence or absence of poly(I:C); error bars indicate standard deviation between mice.
Type 1 IFN is sufficient but not necessary to lead to breakthrough alloimmunization in the setting of KELIg immunoprophylaxis
These data and past studies in our laboratory led us to hypothesize that the IFN-α/β generated by the recipients following poly(I:C) administration was playing a role in the breakthrough alloimmunization observed in the setting of KELIg immunoprophylaxis.
To investigate this, we first tested whether poly(I:C) resulted in breakthrough alloimmunization in the setting of KELIg immunoprophylaxis in animals unable to produce significant amounts of IFN-α/β.10 We have previously shown that animals lacking IFN regulatory factors 3 and 7 (IRF 3/7−/− mice) do not generate significant anti-KEL responses in a type 1 IFN-dependent alloimmunization model.14 However, these mice generate similar levels of anti-KEL antibodies compared with wild-type mice in the setting of a simple KEL RBC transfusion (supplemental Figure 2). We initially completed KELIg experiments in IRF 3/7−/− mice and noted that these animals had a breakthrough anti-KEL alloimmune response after poly(I:C) and KEL RBC transfusion in the setting of KELIg immunoprophylaxis (data not shown). Because some IFN-α/β can still be generated by select cell subsets in IRF3/7−/− mice,32 we repeated these studies using IFR 3/5/7−/− mice that are devoid of IFN production. Despite an inability to generate IFN-α/β, breakthrough anti-KEL alloimmunization still occurred in these IRF 3/5/7−/− mice after poly(I:C) and KEL RBCs in the setting of KELIg (Figure 6A). Of note, the magnitude of the anti-KEL response in the IRF 3/5/7−/− mice was significantly lower than that observed in wild-type animals at all evaluated time points, suggesting that IFN-α/β production following poly(I:C) treatment played a contributing role.
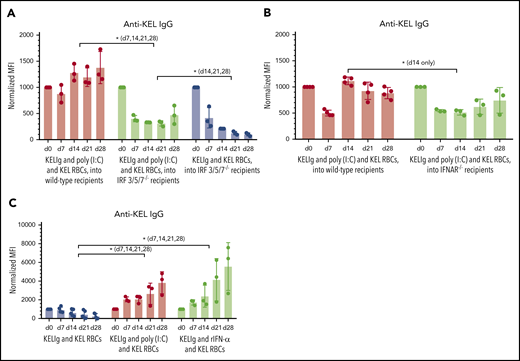
Type 1 IFN is sufficient but not necessary to lead to breakthrough alloimmunization in the setting of KELIg immunoprophylaxis. Total anti-KEL IgG was measured in the serum at the indicated time points posttransfusion (A) in wild-type or IRF 3/5/7−/− recipients passively immunized with KELIg followed by KEL RBCs in the absence or presence of poly(I:C); (B) in wild-type or IFNAR KO recipients passively immunized with KELIg followed by KEL RBCs in the presence of poly(I:C); and (C) in wild-type animals passively immunized with KELIg followed by KEL RBCs or KEL RBCs mixed with exogenous IFN-α. These data are representative of 2 to 3 independent experiments with 3 to 6 mice per group per experiment (in total, 9, 9, and 9 mice were studied across 2 experiments for panel A; 12 and 11 mice were studied across 3 experiments for panel B; 7, 6, and 7 mice were studied across 2 experiments for panel C). *P < .05 between indicated groups at day 7 (d7), 14, 21, and 28 or at described time points; error bars indicate standard deviation between individual mice.
Type 1 IFN is sufficient but not necessary to lead to breakthrough alloimmunization in the setting of KELIg immunoprophylaxis. Total anti-KEL IgG was measured in the serum at the indicated time points posttransfusion (A) in wild-type or IRF 3/5/7−/− recipients passively immunized with KELIg followed by KEL RBCs in the absence or presence of poly(I:C); (B) in wild-type or IFNAR KO recipients passively immunized with KELIg followed by KEL RBCs in the presence of poly(I:C); and (C) in wild-type animals passively immunized with KELIg followed by KEL RBCs or KEL RBCs mixed with exogenous IFN-α. These data are representative of 2 to 3 independent experiments with 3 to 6 mice per group per experiment (in total, 9, 9, and 9 mice were studied across 2 experiments for panel A; 12 and 11 mice were studied across 3 experiments for panel B; 7, 6, and 7 mice were studied across 2 experiments for panel C). *P < .05 between indicated groups at day 7 (d7), 14, 21, and 28 or at described time points; error bars indicate standard deviation between individual mice.
To further investigate the role of IFN-α/β, animals lacking receptors that sense type 1 IFN (IFNAR1−/− mice) were treated with KELIg and KEL RBCs in the presence or absence of poly(I:C). IFNAR1−/− mice generated anti-KEL alloantibodies in the presence of KELIg and poly(I:C), although the magnitude of the response was subtly lower than that of wild-type animals at most time points (Figure 6B).
The data shown in Figure 6A-B are consistent with IFN-α/β playing a role in poly(I:C)-associated breakthrough alloimmunization in the setting of KELIg, but not being solely responsible. To investigate whether type 1 IFN alone was sufficient to cause breakthrough alloimmunization in the setting of KELIg, wild-type recipients were treated with KELIg and cotransfused with exogenous recombinant IFN-α mixed with KEL RBCs. Similar to what was observed with poly(I:C), treatment with exogenous IFN-α led to breakthrough alloimmunization despite KELIg immunoprophylaxis (Figure 6C).
Discussion
Antibody-mediated immunoprophylaxis against the D antigen (RhIg), introduced >50 years ago, changed the landscape of obstetrics and neonatology regarding the risk of hemolytic disease of the fetus and newborn. However, the mechanism of action of RhIg remains elusive, and breakthrough anti-D alloimmunization still occurs. In this study, we describe breakthrough anti-KEL alloimmunization in a murine model despite immunoprophylaxis (ie, immunoprophylaxis failure) in the setting of recipient inflammation. To our knowledge, this is the first time that the impact of antiviral responses on immunoprophylaxis efficacy has been evaluated.
Our most unexpected finding was that the type 1 IFN (IFN-α/β) generated in recipients after poly(I:C) treatment was sufficient but not necessary to lead to immunoprophylaxis failure. We used IRF 3/5/7−/− animals in some experiments, as cells from these animals are incapable of generating IFN-α/β.32 The fact that some degree of breakthrough alloimmunization still occurred in these triple knockout animals despite KELIg immunoprophylaxis in the setting of poly(I:C) led us to conclude that the opsonization of KEL RBCs that results following KELIg administration likely synergizes with the type 1 IFN induced by poly(I:C) to contribute to immunoprophylaxis failure. Poly(I:C) is known to induce NF-κB target genes (eg, IL-6, TNF-α) and IFN-stimulated genes, directly through IRFs as well as indirectly.33,34 Furthermore, the generation of complement breakdown products has been shown to influence pattern recognition receptor signaling in other systems,35 and ongoing studies are investigating the possible role(s) these products may play in our model.
A critical question that remains unanswered is how KELIg prevents alloimmunization. Past work led us to postulate that antigen modulation of the KEL glycoprotein by KELIg was important to its mechanism of action.6 In the current study, transfused RBCs were cleared, and the KEL glycoprotein became undetectable just as quickly in poly(I:C)-treated mice as in animals transfused with KEL RBCs in the absence of inflammation. The clearance findings are consistent with anti-D immunoprophylaxis studies by Kumpel, with no specific RBC clearance patterns identified to predict the efficacy of anti-D monoclonal or polyclonal antibodies.36 They are also consistent with findings by Stegmann et al, which showed a lack of apparent influence of high-affinity alleles encoding FcγRs on RhIg immunoprophylaxis efficacy in humans.28
Despite the lack of a difference in overall peripheral blood KEL RBC clearance patterns, it is possible that the changes in RBC consumption patterns observed by splenic cells in the setting of poly(I:C) play a role in KELIg immunoprophylaxis failure. Our current data suggest a possibility that CD8α DC consumption contributes in part to KEL antigen “ignorance” in the setting of KELIg alone, whereas inflammatory monocyte consumption37 contributes in part to alloimmunization in the setting of KELIg with poly(I:C). Although CD4+ T-cell help is not required for anti-KEL responses following a simple transfusion in this model,25 animals lacking CD4+ T cells have altered alloantibody induction and evanescence kinetics compared with wild-type mice. Furthermore, although CD4+ T-cell help is not required for immunoprophylaxis efficacy at baseline, it remains possible that T-cell activation following cytokine production or complement activation that occurs around the time of or as a result of KEL RBC consumption by splenic subsets may play an important role. Inclusion of poly(I:C) clearly engages a CD4+ T-cell–dependent process,38 illustrating that the mechanisms of alloimmunization that drive anti-KEL antibody formation in this setting may fundamentally differ from what occurs in noninflamed recipients.
Study limitations must always be considered. We focused on recipient inflammation with poly(I:C),16 and findings observed after poly(I:C) or exogenous IFN-α/β administration cannot be extrapolated to all types of inflammation. Our data show that type IFN-α/β generation in response to poly(I:C) is important, yet not completely necessary for breakthrough alloimmunization. As such, additional signaling pathways remain to be identified. The increased IL-6 and MCP-1 levels observed may be contributing factors, with IFN-α/β, IL-6, and MCP-1 being shown in other models to have both interdependent and independent roles in inflammation.39 Of note, a susceptibility to alloimmunization based on inflammatory gene variants40,41 has not yet been explored in this KELIg model. The applicability of our findings to other RBC antigens remains to be determined. For example, although poly(I:C) enhances antibody formation to the HEL antigen,16,42 anti-HEL antibody formation requires CD4+ T cells even under noninflammatory conditions.43 This suggests that while marginal zone B cells and perhaps other early immune regulators may initiate alloimmunization following RBC transfusion,25 downstream immune pathways engaged by distinct alloantigens may differ. Similarly, the consequence of antibody engagement of these RBC antigens may likewise differ.8,23,38,44,45
The translation of these animal model findings to humans remains to be determined. However, it is possible that females with baseline inflammation due to diseases associated with type 1 IFN such as systemic lupus erythematosus or rheumatoid arthritis may be at increased risk of RhIg failure. It is also possible that females with transient inflammation (such as an acute viral infection) or those undergoing IFN-based treatments at the time of RhIg administration may be more likely to have breakthrough anti-D alloimmunization, although no studies to date have investigated these possibilities. Understanding the role of antiviral inflammation in breakthrough alloimmunization could lead to the development of interventions to improve immunoprophylaxis efficacy.
In sum, our data show that breakthrough anti-KEL RBC alloimmunization occurred after poly(I:C) treatment despite KELIg immunoprophylaxis in a murine model. IFN-α administration could recapitulate breakthrough alloimmunization but recipient type 1 sensing was not required for poly(I:C)-induced breakthrough alloimmunization. Although RBC posttransfusion recovery and survival appeared to be similar with or without inflammation, differences in patterns of splenic cell consumption and serum cytokines emerged, with inflammatory monocytes potentially playing an important role. These data increase our understanding of the successes and failures of immunoprophylaxis in mice and provide insight into potential causes of RhIg immunoprophylaxis failure in humans.
For original data, please contact jeanne.hendrickson@yale.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Akiko Iwasaki for experimental input, and Michael Diamond and Sujan Shresta for providing access to the Irf3/5/7−/− animals.
This work was funded by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grants R01 HL126076, R01 HL132951 (J.E.H.), 5 K08 HL141446 (D.R.G.), and P01 HL132819 (J.C.Z.). It was also funded in part by NIH, National Cancer Institute grant P30CA016359 and NIH, National Institute of Diabetes and Digestive and Kidney Diseases grant U54 DK106857 (Yale Cooperative Center of Excellence in Hematology).
Authorship
Contribution: V.E.-R., J.L., D.R.G., J.E.F., S.R.S., K.E.H, and J.E.H. planned the experiments; V.E.-R., J.L., D.R.G., M.S., D.L., and J.E.F. executed most of the experiments; in addition to other authors, C.J.L. and J.C.Z. made experimental suggestions; J.E.H. wrote the initial draft of the manuscript; V.E.-R. generated many of the figures; and all authors edited the manuscript and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeanne E. Hendrickson, Yale University Departments of Laboratory Medicine and Pediatrics, 330 Cedar St, Clinic Building 405, PO Box 208035, New Haven, CT 06520-8035; e-mail: jeanne.hendrickson@yale.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal