

In this issue of Blood, report a new xenograft mouse model with enhanced human hematopoietic and tumor engraftment, created by knocking the human SIRPA gene into immunodeficient mice (see figure).1
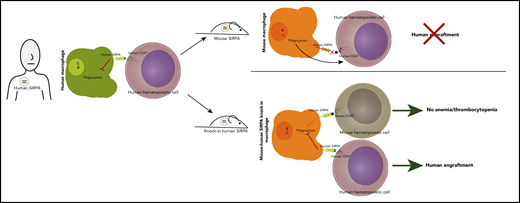
Knock-in of human SIRPA into immunodeficient C57BL/6 recombination activating genes 1/2 (Rag1/2null), interleukin-2 (IL-2) receptor common γ chain subunit (IL2rgnull) strain (BRGS) leads to enhanced human engraftment without inducing anemia/thrombocytopenia.
Knock-in of human SIRPA into immunodeficient C57BL/6 recombination activating genes 1/2 (Rag1/2null), interleukin-2 (IL-2) receptor common γ chain subunit (IL2rgnull) strain (BRGS) leads to enhanced human engraftment without inducing anemia/thrombocytopenia.
The ability of human hematopoietic cells injected into mice to engraft and establish multilineage human hematopoiesis is a remarkable feat that has facilitated seminal discoveries in hematopoiesis, cancer, infectious disease, and immunology.2,3 Thanks to decades of research to identify and overcome barriers to human-mouse engraftment and further optimize murine xenograft strains, numerous robust models are currently available to researchers. However, limitations in some xenograft models still exist, including incomplete myeloid reconstitution, low capacity to support long-term serially transplantable human engraftment, and/or reduced longevity of xenografted animals. Jinnouchi et al sought to address some of these limitations via humanization of the murine phagocytic cells.
Mice can reject human cells via innate and adaptive immune responses, and these murine defenses must be overcome to create robust humanized models. Depletion of T and B cells via either mutation of the Prkdc gene or deletion of Rag1/2null and ablation of NK cell function, most commonly via deletion of IL2rgnull, can prevent rejection of the transplanted human cells. An additional necessity for human-mouse engraftment is so-called phagocytic tolerance, mediated by the signal-regulatory protein α (SIRPA)–CD47 axis.4 SIRPA is a transmembrane protein expressed on macrophages. When SIRPA binds CD47, a ubiquitously expressed cell surface protein, it sends a “do not eat” signal to the macrophage; in its absence, phagocytosis is triggered, and the unrecognized cell is engulfed and destroyed. SIRPA-CD47 binding is generally species specific; however, nonobese diabetic (NOD) and BALBc mice have Sirpa polymorphisms that result in some capacity to bind human CD47. This binding likely explains their superior capacity for human engraftment over other murine strains, including C57BL/6, which has a SIRPA that does not bind to human CD47.4 Numerous strategies to further enhance phagocytic tolerance in immunocompromised mice have somewhat improved human engraftment, including transgenic expression of human SIRPA,5 BAC transgenesis of human SIRPA,6 knock-in of the extracellular CD47-binding portion of human SIRPA into murine Sirpa,7 and knock-in of the NOD Sirpa into the BRGS.
With these findings in mind, Jinnouchi et al postulated that biallelic knock-in of the full human SIRPA gene would further facilitate human engraftment in the BRGS (BRGShuman). Indeed, they found that this strategy allowed for efficient engraftment of human hematopoietic cells in the bone marrow and peripheral blood (see figure). The transplanted BRGShuman mice had high levels of sustained human engraftment even 24 weeks after transplantation and had the capacity to serially transplant, indicative of long-term stem cell self-renewal. The BRGShuman mice also displayed higher myeloid reconstitution compared with many commonly used xenograft murine models that mainly exhibit lymphoid engraftment.3 Because human hematopoiesis is generally myeloid predominant, this model may more faithfully recapitulate normal human hematopoiesis and facilitate study of myeloid disorders.
The authors found that primary samples from acute myeloid leukemia (AML) and colon cancer patients readily engrafted in BRGShuman mice, highlighting the utility of this model in studying both normal hematopoiesis and cancer. Furthermore, the authors showed that BRGShuman mice were far more tolerant of radiation than NSG mice, which are notoriously intolerant of irradiation and chemotherapy. Presumably, the more resilient BRGShuman would also better tolerate cytotoxic chemotherapy and may therefore be better suited for preclinical investigation of anticancer agents.
The authors noted that the BRGShuman mouse was healthy and had a long lifespan, somewhat surprising considering that mice experience anemia/thrombocytopenia after knockout of murine Sirpa, likely because of phagocytosis of red blood cells (RBCs) and platelets. One would presume the BRGShuman mice, which lack any murine SIRPA, might meet the same fate, but the authors found that human SIRPA can bind to C57BL/6 CD47 with sufficient affinity to prevent phagocytosis of critical blood components.
Therefore, the BRGShuman mouse line represents another tool in the toolbox for in vivo investigation of human hematopoiesis and cancer. But where does it fit into the current landscape of murine xenotransplant models? Certainly, other strategies have improved engraftment and enhanced myeloid reconstitution, including expression of human stem/myeloid-supporting cytokines either through transgenic expression (NSG mice transgenically expressing human SCF, granulocyte-macrophage colony-stimulating factor, and IL-3 [NSGS]) or knock-in of human myeloid-supporting cytokines to the endogenous murine loci with or without BAC transgenic expression of human SIRPA (MITRG and MISTRG, respectively).6 Myeloid lineage and AML engraftment are enhanced within the readily available NSGS mice; however, this comes at the expense of stem cell self-renewal capacity, which is poor in this model.2 MISTRG mice show excellent myeloid reconstitution but develop anemia, likely as a result of phagocytosis of mouse and human RBCs.6 Additionally, because of dysfunctional murine hematopoietic stem cells (HSCs) that are outcompeted by the transplanted human HSCs, immunodeficient mice with Kit receptor mutations have the capacity for efficient, multilineage, serially transplantable human engraftment without a requirement for pretransplantation conditioning with radiation or busulfan.8 Although the BRGShuman model overcomes some of these limitations, ultimately combinations of varying murine lines may further improve human-mouse xenografts.
With new anticancer therapies emerging, reliable xenograft models capable of supporting engraftment of all types of malignancies are necessary. Some hematologic malignancies are still associated with poor engraftment, including myelodysplastic syndrome, core-binding factor AML, and myeloproliferative neoplasms. Newer strategies such as knock-in of human SIRPA and human myeloid-supporting cytokines might address these limitations.9 Additionally, long-term xenotransplants of HSCs from individuals with clonal hematopoiesis may deepen our understanding of how and under what circumstances certain HSCs preferentially expand over time. Finally, increasingly scientists are editing the genomes of human HSCs with CRISPR/Cas9, including for potential therapeutic use.10 Accurate animal model systems are essential to ensure that edited human HSCs maintain the capacity to engraft, support long-term multilineage hematopoiesis, and have the intended phenotypic consequence of the engineered genetic change before in-human use. The evolution of murine xenograft models will undoubtedly continue, and these advances will lead to greater understanding of the function of normal, malignant, and edited stem cells.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal