

Key Points
CH, including donor-engrafted CH, is highly prevalent among donors and recipients long-term after allo-HSCT.
CH clones variably expand at different levels of the hematopoietic hierarchy and can clonally evolve into subclones.

Abstract
Clonal hematopoiesis (CH) is associated with age and an increased risk of myeloid malignancies, cardiovascular risk, and all-cause mortality. We tested for CH in a setting where hematopoietic stem cells (HSCs) of the same individual are exposed to different degrees of proliferative stress and environments, ie, in long-term survivors of allogeneic hematopoietic stem cell transplantation (allo-HSCT) and their respective related donors (n = 42 donor-recipient pairs). With a median follow-up time since allo-HSCT of 16 years (range, 10-32 years), we found a total of 35 mutations in 23 out of 84 (27.4%) study participants. Ten out of 42 donors (23.8%) and 13 out of 42 recipients (31%) had CH. CH was associated with older donor and recipient age. We identified 5 cases of donor-engrafted CH, with 1 case progressing into myelodysplastic syndrome in both donor and recipient. Four out of 5 cases showed increased clone size in recipients compared with donors. We further characterized the hematopoietic system in individuals with CH as follows: (1) CH was consistently present in myeloid cells but varied in penetrance in B and T cells; (2) colony-forming units (CFUs) revealed clonal evolution or multiple independent clones in individuals with multiple CH mutations; and (3) telomere shortening determined in granulocytes suggested ∼20 years of added proliferative history of HSCs in recipients compared with their donors, with telomere length in CH vs non-CH CFUs showing varying patterns. This study provides insight into the long-term behavior of the same human HSCs and respective CH development under different proliferative conditions.
Introduction
Clonal hematopoiesis (CH) is defined as the occurrence of recurrent mutations in known oncogenes in hematopoietic stem and progenitor cells (HSPCs) in the absence of overt hematologic malignancies. CH is an age-related1-4 condition and associated with an increased risk of hematological cancers,1,5-7 including therapy-related myeloid malignancies,8,9 cardiovascular disease,1,10,11 thromboembolism,12 and all-cause mortality.1
During allogeneic hematopoietic stem cell transplantation (allo-HSCT), a small percentage of hematopoietic stem cells (HSCs) are transferred from the donor to the recipient. Whereas the hematopoietic system is largely unaffected in the donor with negligible HSC expansion occurring due to HSC donation, transplanted donor HSCs within the recipient undergo substantial self-renewing expansion until homeostatic conditions have been regained (Figure 1A). In addition, the transplant conditioning regimen elicits a highly inflammatory milieu contributing to enhanced HSC cell divisions. Altogether, the increased proliferative history of donor HSCs within the recipient as compared with donor HSCs within the donor leads to a measurable difference in telomere lengths between hematopoietic cells in donors and recipients equivalent to a premature ageing of ∼10 to 30 years.13-15 Thus, we hypothesized that increased HSC proliferation within an inflammatory microenvironment in recipients might promote emergence and/or clonal selection and evolution of CH clones.16 In fact, previous studies have shown that during autologous8,17 and allogeneic18,19 HSCT, preexisting donor CH clones can engraft and clonally evolve within the recipient, occasionally leading to donor cell leukemia.18-20 Notably, CH in the setting of allo-HSCT is an emerging clinical factor to consider with a potential impact on outcome.21 However, more data are needed to better estimate the frequency of CH in the setting of allo-HSCT, especially with a longer follow-up time. In this study, we set out to investigate the potential role of CH in long-term survivors of allo-HSCT and their respective related donors.

Characteristics of somatic mutations found in allo-HSCT cohort. (A) Schematic outline depicting study design and analyses performed. (B) Spectrum of somatic mutations found in donors and recipients of allo-HSCT. (C) Number of individuals with 1, 2, or 3 mutations. (D) Type of mutations. (E) Distribution of types of base pair changes for single-nucleotide variants. (F) VAFs in the total cohort and in donors and recipients, respectively. Mann-Whitney test was used to calculate statistical significance (ns, nonsignificant).
Characteristics of somatic mutations found in allo-HSCT cohort. (A) Schematic outline depicting study design and analyses performed. (B) Spectrum of somatic mutations found in donors and recipients of allo-HSCT. (C) Number of individuals with 1, 2, or 3 mutations. (D) Type of mutations. (E) Distribution of types of base pair changes for single-nucleotide variants. (F) VAFs in the total cohort and in donors and recipients, respectively. Mann-Whitney test was used to calculate statistical significance (ns, nonsignificant).
Study subjects, materials, and methods
Study population
A single-center cohort of n = 45 HSCT recipients and their related donors was evaluated. Forty-two donor-recipient pairs were eventually enrolled in this study after giving informed consent. The study was approved by the local ethics committee (KEK-ZH no. 2015-0053) in accordance with the Declaration of Helsinki.
Cell isolation
Granulocytes were isolated from 10 mL EDTA anticoagulated peripheral blood using EasySep Direct Neutrophil Isolation Kit (StemCell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions.
CD34+ HSPCs were isolated from 20 mL EDTA anticoagulated peripheral blood using human CD34 MicroBead Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s recommendations. B cells, T cells, and monocytes were flow-sorted from CD34− cell fractions using a FACSAria III Flow Cytometer (BD Biosciences, San Jose, CA).
DNA isolation
DNA from granulocytes, monocytes, B cells, and T cells was isolated using QIAamp DNA Mini Kit (Hilden, Germany) according to the manufacturer’s recommendations.
CFU assay
CD34+ HSPCs were plated in 9 mL cytokine-supplemented methylcellulose medium (StemCell Technologies) as described previously.22 After 14 days of culture at 37°C and 5% CO2 single colony-forming units (CFUs) were picked and each resuspended and processed in 20 µL QuickExtract DNA Extraction Solution (Lucigen, Middleton, WI). To detect the genetic mutations of interest in single CFUs, DNA was subjected to polymerase chain reaction (PCR) with primers for each gene followed by Sanger sequencing.
Telomere length analysis
Library preparation for NGS
For next-generation sequencing (NGS) from granulocyte DNA (50 ng per sample), libraries were prepared using the HaloPlex HS Target Enrichment System for Illumina sequencing from Agilent according to the specifications. The libraries were then sequenced using the Illumina HiSeq 2500 System.
For validation NGS from granulocyte, monocyte, B-cell, and T-cell DNA, 200- to 280-bp amplicons were PCR amplified, purified, and submitted for NGS (Massachusetts General Hospital, Center for Computational and Integrative Biology Core Facility, Cambridge, MA).
NGS data processing and analysis
Adapter contamination and low-quality bases were removed from the raw reads using SeqPurge27 (git commit id 71885f4). The reads were aligned to the human reference genome hg19 using bwa mem (v0.7.15).28 Picard tools (v2.8.3) were then used to fix erroneous mate information as well as sort and merge the alignment files for each sample. All alignments marked as secondary were removed before performing an indel realignment using GATK (3.8).29 In the next step, unique molecular identifier (UMI) clusters were identified with UMI-tools (v0.4.4),30 and sequencing errors were removed by generating the consensus reads for every biological template using an in-house tool. The resulting BAM files were processed with samtools mpileup (v1.3.1).31 Next, VarScan2 (v2.4.3)32 was used to call single-nucleotide variants and indels with the command mpileup2snp and mpileup2indel. The VarScan2 results were combined for each sample and annotated with dbSNP (version 138)33 and cosmic (version 80)34 using SnpSift (v4_3p_core)35 before using SnpEff (v4_3p_core)36 for the final annotation. Expert manual curation of the identified mutations with regards to biological impact defined a set of mutations that were investigated using IGV (2.3.68)37 and validated using ultra-deep amplicon sequencing. For validation, the reads were mapped, sorted, and realigned using the same specifications as above. The resulting alignments were then processed with samtools mpileup, and variants were called using VarScan2 using the same parameters as described above.
Statistical analysis
Significance of differences was analyzed using nonparametric tests as indicated in the figure legends. A difference between groups was considered statistically significant if values were P < .05. Statistical analysis was calculated using Prism software.
Results
Somatic mutations in donors and recipients of related allo-HSCT
Based on long-term survival defined as ≥10 years after allo-HSCT, we evaluated a cohort of recipients (n = 45) and their respective sibling donors (n = 45) who underwent related allo-HSCT at our institution between 1983 and 2006 (Figure 1A). Three allo-HSCT donor-recipient pairs were excluded from the analyses for reasons described in greater detail below. Forty-two allo-HSCT donor-recipient pairs (ie, 84 individuals) were eventually included in the analysis. The median age at transplantation in the total cohort, donors, and recipients was 38, 37, and 39 years, respectively (Table 1). The median age at study inclusion was 57 years (range, 29-95 years) for donors and 61 years (range: 32-77 years) for recipients, which was not significantly different. The median follow-up time since transplantation was 16 years (range, 10-32 years). The gender distribution was balanced, and most patients were transplanted for malignant disease. Twenty-eight patients received bone marrow (BM), while 14 patients were transplanted with peripheral blood (PB) stem cells (Table 1). We collected PB from study participants and isolated DNA from purified populations of granulocytes, monocytes, B and T cells. In addition, PB CD34+ HSPCs were isolated and used for clonogenic CFU assays followed by colony picking and genomic DNA extraction. The resulting DNA from these various sources were subjected to several downstream applications that included targeted NGS with increased sensitivity due to the use of UMIs (supplemental Table 1, available on the Blood Web site), Sanger sequencing, and quantitative PCR (Figure 1A).
Demographic and clinical annotation of study cohort
| Variable | Study participants (n = 84) | Donors (n = 42) | Recipients (n = 42) |
|---|---|---|---|
| Patient-related variables | |||
| Age (y), median (range) | |||
| At transplantation / donation | 38 (15-65) | 37 (15-65) | 39 (16-58) |
| At study inclusion | 59 (29-95) | 57 (29-95) | 61 (32-77) |
| Time since transplantation (y), median (range) | 16 (10-32) | ||
| Gender | |||
| Male | 45 | 22 | 23 |
| Female | 39 | 20 | 19 |
| Disease | |||
| AML | 17 | ||
| MDS | 1 | ||
| CML | 6 | ||
| ALL | 8 | ||
| NHL | 3 | ||
| MM | 2 | ||
| SAA | 2 | ||
| Nonmalignant | 3 | ||
| Transplantation-related variable | |||
| Graft type | |||
| PBSCs | 14 | ||
| BM | 28 |
| Variable | Study participants (n = 84) | Donors (n = 42) | Recipients (n = 42) |
|---|---|---|---|
| Patient-related variables | |||
| Age (y), median (range) | |||
| At transplantation / donation | 38 (15-65) | 37 (15-65) | 39 (16-58) |
| At study inclusion | 59 (29-95) | 57 (29-95) | 61 (32-77) |
| Time since transplantation (y), median (range) | 16 (10-32) | ||
| Gender | |||
| Male | 45 | 22 | 23 |
| Female | 39 | 20 | 19 |
| Disease | |||
| AML | 17 | ||
| MDS | 1 | ||
| CML | 6 | ||
| ALL | 8 | ||
| NHL | 3 | ||
| MM | 2 | ||
| SAA | 2 | ||
| Nonmalignant | 3 | ||
| Transplantation-related variable | |||
| Graft type | |||
| PBSCs | 14 | ||
| BM | 28 |
AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; PBSCs, peripheral blood stem cells; SAA, severe aplastic anemia.
With a mean coverage of 582 after consensus computation and a variant allele frequency (VAF) cutoff set at 0.01, we detected 35 mutations in 11 genes in granulocyte DNA (Figure 1B and supplemental Table 2). Notably, all mutations reported in this study were independently validated by ultra-deep (coverage 50 000-100 000×) amplicon NGS with a high degree of concordance between VAFs determined by targeted NGS and ultra-deep amplicon NGS (supplemental Figure 1). In line with previous studies on CH, DNMT3A and TET2 were the most frequently mutated genes, together accounting for 57.1% of all mutations.38 We did not detect ASXL1 mutations in our cohort, which was likely due to statistical chance and/or the highly selected patient cohort and the relatively young median age in our cohort. Validation experiments demonstrated that our sequencing and analysis pipeline was capable of robustly detecting ASXL1 variants in clinical samples from patients with myeloid malignancies (supplemental Figure 2). The remaining mutations affected genes commonly mutated in CH and myeloid malignancies. There was no clear overrepresentation of specific mutations in either donors or recipients (Figure 1B). More than 1 mutation (up to 3 mutations) per individual could be found in 11 study participants (Figure 1C). Most mutations (18 mutations) were missense mutations, followed by indels and nonsense and splice mutations (Figure 1D). The most frequent single-base substitutions were cytosine-to-thymine (C→T) transitions known to be associated with ageing39,40 (Figure 1E). The median VAF in the total cohort was 0.03, with no statistical difference between donors and recipients (Figure 1F).
Altogether, we detected CH in a total of 23 out of 84 study participants corresponding to 27.4% of the total cohort (Figure 2A). At least 1 mutation was found in 10 out of 42 donors (23.8%) and 13 out of 42 recipients (31%), but this difference was not statistically significant. Individuals with CH were significantly older than study participants without CH when analyzed for age at study inclusion (Figure 2B) as well as age at transplantation (supplemental Figure 3). This well-established age association of CH was also found when donors and recipients were analyzed separately (Figure 2B; supplemental Figure 3). Notably, there was no association between the magnitude of VAFs and time since transplantation (data not shown).
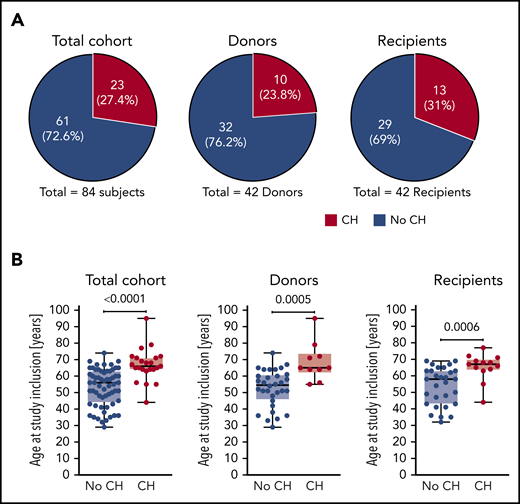
CH in donors and recipients of allo-HSCT. (A) Proportion of study participants affected by CH in the total cohort, donors, and recipients, respectively. (B) Age at study inclusion of study participants affected by CH in the total cohort, donors, and recipients, respectively. Mann-Whitney test was used to calculate statistical significance.
CH in donors and recipients of allo-HSCT. (A) Proportion of study participants affected by CH in the total cohort, donors, and recipients, respectively. (B) Age at study inclusion of study participants affected by CH in the total cohort, donors, and recipients, respectively. Mann-Whitney test was used to calculate statistical significance.
Donor-engrafted CH in recipients of related allo-HSCT
Based on the detection of ≥1 identical (often 2 identical) nonhotspot mutations in both the donor and recipient, we could identify several cases of donor-engrafted CH (Figure 3A-C); ie, a scenario where a preexisting CH clone was transferred at the time of transplant from the donor and engrafted in the recipient. Altogether, donor-engrafted CH occurred in 5 out of 42 transplantations (11.9%) as well as in 5 out of 10 (50%) and in 5 out of 13 (38.5%) donors and recipients with CH, respectively (Figures 2A and 3A). Of note, 1 case of donor-engrafted CH eventually progressed to MDS. The corresponding mutational data were, however, analyzed separately as discussed below. In all other cases of donor-engrafted CH, VAFs in the recipient increased significantly relative to the corresponding donor over a long period of time (median time since transplant, 15 years) (Figure 3B-D). Notably, these donors were significantly older, with a median age at transplantation of 50 years compared with a median donor age at transplantation of 37 years in the entire cohort (Figure 3D and Table 1) and 35 years in the subgroup of donors that did not result in donor-engrafted CH (Figure 3D). These findings together with the well-established age association of CH strongly suggest that an increased donor age at transplantation may be associated with an increased risk of donor-engrafted CH.

Donor-engrafted CH in allo-HSCT. (A) Proportion of donor-recipient pairs affected by donor-engrafted CH. (B) VAFs in individual donor-recipient pairs of cases of donor-engrafted CH. (C) Pooled analyses of VAFs in donor-recipient pairs with of donor-engrafted CH. Wilcoxon matched-pairs signed rank test was used to calculate statistical significance. (D) Donor age at transplantation for cases of donor-engrafted CH. Mann-Whitney test was used to calculate statistical significance. (E) Case example of donor-engrafted CH progressing to myelodysplastic syndromes. D, donor; R, recipient.
Donor-engrafted CH in allo-HSCT. (A) Proportion of donor-recipient pairs affected by donor-engrafted CH. (B) VAFs in individual donor-recipient pairs of cases of donor-engrafted CH. (C) Pooled analyses of VAFs in donor-recipient pairs with of donor-engrafted CH. Wilcoxon matched-pairs signed rank test was used to calculate statistical significance. (D) Donor age at transplantation for cases of donor-engrafted CH. Mann-Whitney test was used to calculate statistical significance. (E) Case example of donor-engrafted CH progressing to myelodysplastic syndromes. D, donor; R, recipient.
Besides these 5 cases of donor-engrafted CH, we identified 5 cases of donor-only CH and 8 cases of recipient-only CH. To rule out that in the latter cases of recipient-only CH the presence of clones is not due to residual recipient hematopoiesis but developed from the grafted donor-derived hematopoiesis, we performed chimerism analysis using digital PCR. While most cases of recipient-only CH showed 100% donor chimerism (supplemental Figure 4 and supplemental Table 3), we identified 2 recipients (R30 and R33) originally transplanted for severe aplastic anemia with 0% donor chimerism and 1 recipient (R16) transplanted for CML with ∼10% residual recipient hematopoiesis that was, of note, BCR-ABL1+. These 3 donor-recipient pairs were therefore excluded from our initial cohort of 45 donor-recipient pairs as mentioned above.
Furthermore, 1 of the 5 donor-engrafted CH cases progressed into MDS in both the donor and the recipient (Figure 3E; supplemental Table 4). We could rule out the possibility of an underlying known genetic predisposition by sequencing the donor and recipient’s DNA using a panel of genes commonly mutated in inherited BM failure syndromes (supplemental Table 5). NGS of the donor and recipient’s granulocyte DNA at the time of study inclusion in 2017 allowed us to retrospectively decipher and reconstruct the origin and clonal evolution of CH progressing to phenotypically different MDS in this donor-recipient pair. A relatively complex mutational pattern in both individuals each carrying 4 mutations at varying VAFs could be revealed (Figure 3E; supplemental Table 4). Importantly, 2 identical mutations were shared between the siblings, demonstrating that the TET2R550* / U2AF1Q157P double-mutant founding clone must have been transferred from the donor to the recipient in 1996 at the time of transplantation. Thereafter, both clones followed different clonal trajectories. In the donor, the TET2R550*/U2AF1Q157P double-mutant founding clone acquired an ASXL1Q760* and a second TET2 mutation (Q967*). In the male recipient, the donor-derived founding clone acquired an X-chromosomal STAG2R259* mutation that has exerted a strong selective advantage, since it (based on VAFs) completely outcompeted the donor-derived TET2R550*/U2AF1Q157P double-mutant founding clone and virtually any residual normal cells. In addition, there was a smaller subclone carrying a SETBP1G870S mutation detectable in the recipient.
Quantitative representation of clones at different levels of the hematopoietic hierarchy
Knowledge of CH stems largely from NGS of PB mononuclear cells (PBMCs). Few studies have investigated the relative clonal expansion of CH mutations in various mature blood cell lineages such as granulocytes, monocytes, and B and T lymphocytes.41-43 Therefore, we addressed the question whether there are distinct representation patterns of CH clones at different levels of the hematopoietic hierarchy and within different hematopoietic lineages. To this end, we performed CFU assays from PB CD34+ cells isolated from study participants with CH and analyzed mutations by Sanger sequencing. We also fluorescence-activated cell sorter–purified monocytes, B cells, and T cells and performed targeted amplicon deep sequencing.
All types of mutations (ie, missense, nonsense, or frameshift) could reliably be identified by Sanger sequencing, and as expected, all mutations were heterozygous (or hemizygous in the case of X-chromosomal mutations in males), as exemplified by some of the resulting Sanger electropherograms (Figure 4A). There was a good correlation between the percentage of detected mutation-positive CFUs by Sanger sequencing and the percentage of predicted mutation-positive CFUs based on VAFs in granulocytes. However, there was a case (TET2S1668fs) that showed a substantial deviation from the expected representation (Figure 4B). This finding of disproportionate clonal representation at the level of HSPCs compared with mature granulocytes suggests that some mutations, besides promoting expansion of HSPCs, may either confer a concomitant lineage differentiation defect or, alternatively, a reduced half-life of the mature progeny. Alternatively, the capacity to form colonies and grow in methylcellulose might be influenced by CH driver mutations.

Quantitative representation of CH clones at different levels of the hematopoietic hierarchy. (A) Sanger sequencing electropherograms exemplifying detection of missense, nonsense, and frameshift mutations in individual CFUs from PB CD34+ cells. (B) Correlation between the percentage of detected mutation-positive CFUs by Sanger and the percentage of predicted mutation-positive CFUs based on VAFs. (C) VAFs in granulocytes, monocytes, B cells, and T cells in donors and recipients shown as a heatmap. (D) VAFs in granulocytes, monocytes, B cells, and T cells in donors and recipients (box and whisker plot). Wilcoxon matched-pairs signed rank test was used to calculate statistical significance.
Quantitative representation of CH clones at different levels of the hematopoietic hierarchy. (A) Sanger sequencing electropherograms exemplifying detection of missense, nonsense, and frameshift mutations in individual CFUs from PB CD34+ cells. (B) Correlation between the percentage of detected mutation-positive CFUs by Sanger and the percentage of predicted mutation-positive CFUs based on VAFs. (C) VAFs in granulocytes, monocytes, B cells, and T cells in donors and recipients shown as a heatmap. (D) VAFs in granulocytes, monocytes, B cells, and T cells in donors and recipients (box and whisker plot). Wilcoxon matched-pairs signed rank test was used to calculate statistical significance.
Interestingly, determining VAFs in granulocytes, monocytes, B cells, and T cells individually in donors and recipients, respectively, revealed all possible combinations: uni-, bi-, tri-, and multilineage penetration of mutations (Figure 4C). While mutations with lower VAFs were often found in myeloid cells (granulocytes and/or monocytes), but not in lymphocytes (B and/or T cells), mutations with higher VAFs had a tendency to fully penetrate all mature lineages (Figure 4C). Of note, VAFs in granulocytes and monocytes were significantly higher than in lymphoid cells, with the lowest VAFs seen in T cells (Figure 4D). In the 5 cases of donor-engrafted CH, the pattern and extent of lineage penetration was not different in donors compared with recipients (Figure 4C).
Clonal architecture in individuals carrying multiple CH mutations
About 50% of individuals with CH in our cohort carried 2 or 3 mutations (Figure 1C). Two distinct scenarios with regards to clonal architecture are possible: (1) subclonality, where a founding clone evolves through acquisition of a second mutation; or (2) unrelated clones with independent acquisition of individual mutations in 2 distinct cells. This distinction is of probable clinical importance, as it may be associated with different risks for progression to myeloid neoplasms. Our data from Sanger sequencing of single cell–derived CFUs from PB CD34+ cells allowed us to reconstruct the clonal architecture for a number of individuals in our cohort with >1 mutation.
In fact, we found evidence for both scenarios: subclonality, as indicated by the concomitant presence of 2 different mutations in the same CFU (Figure 5A), and un-related clones, as demonstrated by exclusive detection of only 1 of the 2 individual mutations per CFU (Figure 5B).
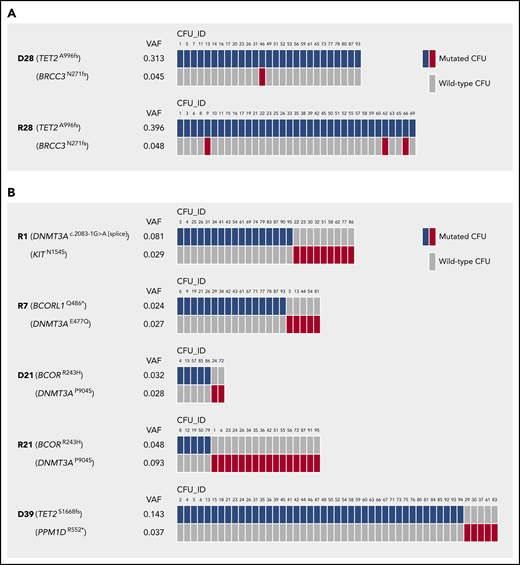
Clonal architecture in individuals carrying >1 CH mutation. (A) Mutations in individual CFUs demonstrating cases of CH with subclonality. (B) Mutations in individual CFUs indicating cases of CH with independent clones. n = 95 single CFUs per individual affected by CH were analyzed for the presence of mutations by Sanger sequencing. Only mutated CFUs are depicted.
Clonal architecture in individuals carrying >1 CH mutation. (A) Mutations in individual CFUs demonstrating cases of CH with subclonality. (B) Mutations in individual CFUs indicating cases of CH with independent clones. n = 95 single CFUs per individual affected by CH were analyzed for the presence of mutations by Sanger sequencing. Only mutated CFUs are depicted.
Collectively, our findings underscore the fact that the clonal architecture, especially in cases with relative low VAFs, cannot be inferred from VAFs alone but requires clonogenic assays.
Telomere length in study participants and individual CFUs from CH carriers
Due to the nature of eukaryotic DNA replication, each cell division leads to a measurable telomere length shortening of ∼50 to 200 bp, which can serve as a molecular counter for the proliferative history of dividing cells and tissues.44,45 Therefore, we determined telomere length in granulocytes by quantitative PCR and found significant telomere length shortening in allo-HSCT recipients as compared with donors (Figure 6A), confirming previous reports demonstrating that the enhanced HSC proliferation occurring in the recipient as a consequence of engraftment and hematopoietic reconstitution during the first year following allo-HSCT indeed leads to a significant telomere length shortening relative to the donor.13-15 The difference in telomere length between donor and recipient translates into ∼20 years of premature ageing of the recipient’s hematopoietic system as compared with the donor’s (Figure 6B).
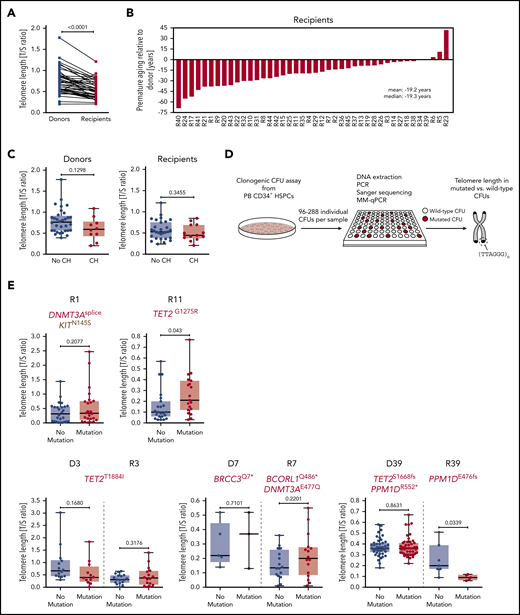
Telomere length in study participants and individual CFUs from CH carriers. (A) Telomere length in donors and recipients, respectively, as measured by MM-qPCR and depicted as T/S ratio. (B) Telomere length difference between individual donor-recipient pairs translated into years of premature ageing. (C) Telomere length in individuals with or without CH based on donor or recipient status. (D) Schematic outline demonstrating experimental workflow for identification of wild-type and mutated CFUs by Sanger sequencing followed by telomere length measurement using MM-qPCR. (E) Telomere length in individual wild-type or CH mutation–positive CFUs (single individuals affected by CH, upper panel; donor-recipient pairs with mutations in both donor and recipient, lower panel). Mann-Whitney test was used to calculate statistical significance.
Telomere length in study participants and individual CFUs from CH carriers. (A) Telomere length in donors and recipients, respectively, as measured by MM-qPCR and depicted as T/S ratio. (B) Telomere length difference between individual donor-recipient pairs translated into years of premature ageing. (C) Telomere length in individuals with or without CH based on donor or recipient status. (D) Schematic outline demonstrating experimental workflow for identification of wild-type and mutated CFUs by Sanger sequencing followed by telomere length measurement using MM-qPCR. (E) Telomere length in individual wild-type or CH mutation–positive CFUs (single individuals affected by CH, upper panel; donor-recipient pairs with mutations in both donor and recipient, lower panel). Mann-Whitney test was used to calculate statistical significance.
We hypothesized that the increased proliferative history of expanded CH clones in either donors or recipients may also lead to a measurable decrease in granulocyte telomere length. There was a trend toward shorter telomeres in donors with CH as compared with donors without CH that was most likely due to the increased age of donors with CH relative to donors without CH. However, individuals with CH did not have significantly shorter telomeres as compared with individuals without CH (Figure 6C). Since this putative association might be masked by (1) the quantitative heterogeneity of telomere lengths between individuals and (2) the relative small sizes of the CH clones in affected individuals, we performed quantitative PCR on DNA from clonogenic CFUs derived from PB CD34+ cells to assess telomere lengths in HSPC-derived myeloid cells from CH mutation–positive and wild-type CFUs (Figure 6D). Intriguingly, these analyses revealed that telomere length did not significantly differ between CH mutation–positive CFUs and wild-type CFUs for most of the analyzed cases, with the exception of 1 case with significantly longer telomeres (Figure 6E, upper panel, R11) and 1 case of significantly shorter telomeres (Figure 6E, lower panel, R39) in CH mutation–positive CFUs. These findings may suggest that HSCs carrying particular CH mutations may activate the canonical or alternative molecular machinery to maintain telomeres in order to prevent telomere attrition. This should be clarified in future studies.
Discussion
In this study, we addressed the question of whether the enforced HSC proliferation during allo-HSCT in conjunction with an inflammatory milieu elicited by the conditioning regimen and posttransplantation situation might promote initiation, expansion, and/or evolution of CH in allo-HSCT recipients as compared with donors. We analyzed a highly selected cohort of relatively young (median age at transplantation/donation, 38 years), related donor-recipient sibling pairs with a long median follow-up (median, 16 years; range, 10-32 years). Thus, this cohort has some inherent selection biases, as discussed below. We used a targeted and error-corrected (ie, UMIs) sequencing approach that allowed us to confidently lower the VAF cutoff for mutation calling to 0.01. We found a high frequency of CH in both donors (23.8%) and recipients (31%), respectively, despite a relatively young median age in our cohort. However, the difference in CH frequency observed between donors and recipients was not statistically significant. In addition, we observed a high percentage of donor-engrafted CH, with 5 out of 42 transplantations (11.9%) resulting in transfer of preexisting CH clones from the donor to the recipient. Donor age at transplantation was significantly higher in the transplantations that resulted in donor-engrafted CH as compared with the transplantations that did not lead to donor-engrafted CH. This finding suggests that donor age at transplantation poses a risk factor for donor-engrafted CH. Notably, there was a consistent and significant increase in clone size as measured by VAF in recipients as compared with donors in the cases of donor-engrafted CH. However, this increase in VAFs was only relatively modest (ie, 2.3-fold in median VAF between donors and recipients, with VAFs in most recipients being ∼0.1), indicating that in recipients with donor-engrafted CH, the hematopoietic system is not dominated by a single clone but still largely polyclonal. These findings suggest that allo-HSCT does not provide an exceedingly “fertile soil” for expansion of preexisting CH clones and that the presence of CH in allo-HSCT recipients is, in principle, compatible with long-term survival. A similar conclusion was reached in a recent study assessing CH in the setting of allo-HSCT.21 However, these can only be preliminary conclusions given the inherent survival bias in our cohort of retrospectively analyzed long-term survivors of allo-HSCT. Future studies are needed to systematically and prospectively investigate the potential impact of CH, including donor-engrafted CH, on the outcome of allo-HSCT.
Furthermore, our findings from characterizing the hematopoietic system of individuals with CH will likely stimulate further research, especially regarding progression from premalignant CH to full-blown myeloid malignancy. A surprisingly high percentage (about 50%) of study participants in our cohort had ≥1 mutation. Recently published studies have shown that the likelihood of CH progressing into AML is higher in individuals carrying more mutations.5,6 However, that association was not perfect, as many individuals in the group who remained free of AML still had ≥1 mutation. Our data from sequencing single CFUs of individuals with ≥1 mutation allowing us to distinguish CH cases with clonal evolution (subclonality) from those with multiple independent clones might provide a framework to refine the association between the number of driver mutations per individual and the risk of progression into AML. While subclonality leads to fewer clonal cells but accumulation of multiple mutations within the same subclone, unrelated clones lead to more clonal cells but less mutational burden per clone. It is tempting to speculate that the presence of clonal evolution (ie, higher mutational burden per clone) is a better predictor of the risk of malignant transformation than the actual number of driver mutations per individual.
Along similar lines, higher VAFs, as measured in PBMCs, are generally associated with a higher risk of AML transformation.5,6 However, we observed a case with disproportionate clonal representation when comparing VAFs in granulocytes and percentage of mutation-positive CFUs. While it is possible that driver mutations may only enhance the ability to form CFUs in vitro, it is tempting to speculate that the mutational burden within HSPCs may be underestimated by measuring VAFs in granulocyte or PBMC DNA only. Mechanistically, some mutations, despite inducing expansion on the HSPC level, may also confer a lineage maturation defect during the transition from HSPCs to mature progeny. Alternatively, mutations could also lead to an increased turnover of mature progeny via a propensity to undergo apoptosis. Either scenario might be associated with a different risk for malignant progression. Future mechanistic studies are therefore needed to further substantiate these findings.
We determined the lineage penetration of CH mutations in fluorescence-activated cell sorter–purified granulocytes, monocytes, and B and T cells. The extent of lineage penetration (uni-, bi-, tri-, and multilineage penetration) was different between samples, with multilineage penetration occurring more often in samples with higher VAFs. Consistent with previous findings,42,43 we observed significantly higher VAFs in myeloid cells (ie, granulocytes and monocytes) relative to B and T lymphocytes, whereas another recent study had reported equal VAFs in myeloid and lymphoid compartments.41
Unexpectedly, we observed telomere length maintenance in all but one of the analyzed individuals with CH when comparing telomere lengths between single CFUs carrying mutations and wild-type CFUs. The importance of this intriguing finding remains to be determined in future studies. Clonal expansion of HSPCs can be driven either by increased survival without concomitantly enhanced proliferation or by increased proliferation. While the former scenario does not require telomere maintenance, in the latter scenario of increased proliferation, telomeres may need to be actively maintained to prevent telomere attrition. Again, it is tempting to speculate that different driver mutations might have distinct requirements either for telomerase complex activity or alternative mechanisms of telomere lengthening resulting in a variable risk of disease progression. Accordingly, a recent study found that loss of Dnmt3a, the most commonly mutated gene in CH, in murine HSPCs leads to their immortalization and indefinite transplantability in serial transplantation assays in mice.46 Given that telomere shortening limits HSPC transplantability,47 in conjunction with findings that Dnmt3a regulates telomere biology,48 our data provide the first but still speculative hint that at least in the case of DNMT3A-mutant CH, the telomere maintenance machinery or alternative mechanisms of telomere lengthening might become activated to prevent telomere attrition.
In summary, our analysis of a cohort of long-term survivors of allo-HSCT and their sibling donors demonstrates that CH is a highly prevalent condition in both donors and recipients of allo-HSCT. These findings should prompt not only future clinical trials prospectively investigating its clinical impact on the outcome of allo-HSCT but also basic studies elucidating HSPC biology and pathomechanisms of leukemic transformation.
Note added in proof
Two recent studies by Fabre et al49 and Hansen et al50 investigated CH in elderly twins and found little to no evidence for a genetic predisposition for the development of CH.
The data reported in this article are available in the National Center for Biotechnology Information’s Sequence Read Archive under accession number PRJNA612593.
Presented in oral form as a late-breaking abstract at the 23rd annual meeting of the European Hematology Association, Stockholm, Sweden, 17 June 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by research grants from the Swiss National Science Foundation (310030B_166673/1) and the Clinical Research Priority Program Human Hemato-Lymphoid Diseases of the University of Zurich (M.G.M.), a SystemsX-StemSysMed grant (M.G.M. and R.C.S.), an ERC Synergy grant (609883) (N.B.), and fellowships from the Swiss Cancer League (KLS-3625-02-2015) and the Swiss National Science Foundation (P300PB_161026/1 and P400PM_183862) (S.B.).
Authorship
Contribution: S.B. and C.M.W. devised, performed, and analyzed experiments and wrote the manuscript; J.S. analyzed experiments and wrote the manuscript; F.B., M.S.V.F., E.G., and E.B. performed experiments; U.S., R.C.S., B.L.E., B.M.F., C.G., C.B., N.B., and T.H.B. devised experiments and discussed data; and M.G.M. directed the study and wrote the manuscript.
Conflict-of-interest disclosure: B.L.E. has received research funding from Celgene and Deerfield. He has received consulting fees from GRAIL, and he serves on the scientific advisory boards for and holds equity in Skyhawk Therapeutics and Exo Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Markus G. Manz, Department of Medical Oncology and Hematology, University of Zurich and University Hospital Zurich, Raemistrasse 100, CH-8091 Zurich, Switzerland; e-mail: markus.manz@usz.ch; and Steffen Boettcher, Department of Medical Oncology and Hematology, University of Zurich and University Hospital Zurich, Raemistrasse 100, CH-8091 Zurich, Switzerland; e-mail: steffen.boettcher@usz.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal