

Key Points
FLT3 inhibition induces HDAC8 upregulation through FOXO1- and FOXO3-mediated transactivation in FLT3-ITD+ AML.
Targeting HDAC8 activates p53 and enhances FLT3 TKI–mediated elimination of FLT3-ITD+ AML.

Abstract
Internal tandem duplication (ITD) mutations within the FMS-like receptor tyrosine kinase-3 (FLT3) can be found in up to 25% to 30% of acute myeloid leukemia (AML) patients and confer a poor prognosis. Although FLT3 tyrosine kinase inhibitors (TKIs) have shown clinical responses, they cannot eliminate primitive FLT3-ITD+ AML cells, which are potential sources of relapse. Therefore, elucidating the mechanisms underlying FLT3-ITD+ AML maintenance and drug resistance is essential to develop novel effective treatment strategies. Here, we demonstrate that FLT3 inhibition induces histone deacetylase 8 (HDAC8) upregulation through FOXO1- and FOXO3-mediated transactivation in FLT3-ITD+ AML cells. Upregulated HDAC8 deacetylates and inactivates p53, leading to leukemia maintenance and drug resistance upon TKI treatment. Genetic or pharmacological inhibition of HDAC8 reactivates p53, abrogates leukemia maintenance, and significantly enhances TKI-mediated elimination of FLT3-ITD+ AML cells. Importantly, in FLT3-ITD+ AML patient–derived xenograft models, the combination of FLT3 TKI (AC220) and an HDAC8 inhibitor (22d) significantly inhibits leukemia progression and effectively reduces primitive FLT3-ITD+ AML cells. Moreover, we extend these findings to an AML subtype harboring another tyrosine kinase–activating mutation. In conclusion, our study demonstrates that HDAC8 upregulation is an important mechanism to resist TKIs and promote leukemia maintenance and suggests that combining HDAC8 inhibition with TKI treatment could be a promising strategy to treat FLT3-ITD+ AML and other tyrosine kinase mutation–harboring leukemias.
Introduction
Acute myeloid leukemia (AML) is a highly heterogeneous disease at the clinical and molecular levels. Recent large-scale genomic sequencing efforts have helped to categorize AML into different subtypes based on the mutation profile and its putative impact on AML pathogenesis and prognosis.1 Among these recurrent mutations, internal tandem duplications (ITDs) of the FMS-like receptor tyrosine kinase 3 (FLT3) are the most frequent, presenting in 25% to 30% AML patients.2 This mutation results in constitutive FLT3 phosphorylation and activation of survival/proliferation downstream signaling pathways and confers an increased risk for relapse and a poor prognosis.1,3 Based on its tyrosine kinase characteristic and clinical significance, several FLT3 tyrosine kinase inhibitors (TKIs), such as midostaurin, gilteritinib, and quizartinib, have been approved for targeted therapy in clinical usage. However, when used as single agents, they only partially inhibit the growth of AML cells and result in a transient clinical response,4,5 suggesting the existence of drug-resistance mechanisms. Indeed, secondary mutations in the FLT3 tyrosine kinase domain have been proposed as a frequent mechanism of resistance6 ; however, more recently, mutational analysis of patient samples following TKI treatment revealed other diverse molecular mechanisms in clinical resistance.7,8 Several preclinical studies also suggested that cellular adaptive mechanisms might contribute to TKI resistance.9,10 Therefore, a better understanding of molecular events contributing to drug resistance would aid in the development of strategies to achieve sustained remission.
In a variety of human cancers, histone deacetylase (HDAC) function and/or expression is perturbed and is associated with poor prognosis.11 HDACs regulate gene expression through histone modification and chromatin remodeling, as well as modulate the function of nonhistone proteins through posttranslational modification. Therefore, aberrant activity of HDACs can cause deregulated gene expression and protein function, which may serve as important mechanisms to promote tumorigenesis and drug resistance. In AML, oncogenic fusion proteins can recruit HDACs to certain gene loci to drive leukemogenesis.12 PML-RARα and CBFβ-SMMHC fusion proteins recruit HDACs to suppress the acetylation and activation of p53, thereby promoting the maintenance of leukemia.13,14 HDACs also regulate genome stability by facilitating DNA damage repair,15 and overexpressed HDACs have been implicated in protecting cancer cells from genotoxic insults.16 For example, HDAC3 participates in genome stability maintenance and enhances the DNA damage repair capability of leukemia cells through activating AKT, thereby protecting leukemia cells from chemotoxicity.17-19 Based on the important roles of HDACs in leukemia maintenance and drug resistance, many HDAC inhibitors have demonstrated potent antitumor effects. For instance, the combination of LBH589 plus imatinib could eradicate TKI-resistant chronic myeloid leukemia stem cells (LSCs).20 In FLT3-ITD+ AML, the class III HDAC SIRT1 was specifically upregulated in LSCs. Inhibition of SIRT1 reduced the growth of FLT3-ITD+ AML LSCs and significantly enhanced TKI-mediated killing of leukemia cells.21 Currently, many HDAC inhibitors are in various stages of clinical trials.22 However, because of pan-HDAC inhibition activity, these inhibitors also demonstrated high toxicity, which restricted their clinical usage. Therefore, it is rational to design isoform-specific HDAC inhibitors to eliminate leukemia effectively and minimize the toxicity, which means that the functions of specific HDACs in specific subtypes of leukemia must be elucidated.
Here, we found that HDAC8 was upregulated upon FLT3 inhibition through FOXO1- and FOXO3-mediated transactivation in FLT3-ITD+ AML cells. Upregulated HDAC8 could deacetylate and inactivate p53, leading to leukemia maintenance and drug resistance upon TKI treatment. Inhibition of HDAC8 significantly enhanced the leukemia-killing effect of TKIs, as well as greatly reduced the frequency of primitive FLT3-ITD+ AML cells in a patient-derived xenograft (PDX) leukemia model. We proposed that simultaneously targeting FLT3 and HDAC8 may be a promising strategy to improve the treatment outcome of FLT3-ITD+ AML.
Methods
Cell lines
MV4-11, MOLM-13, Kasumi-1, THP-1, U937, HL-60, NB4, and OCI-AML3 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and antibiotics. HEK293T cells were cultured in Dulbecco’s modified Eagle medium with 10% fetal bovine serum, 2 mM l-glutamine, and antibiotics. Cells were grown at 37°C in a humidified atmosphere containing 5% CO2.
Patient samples
FLT3-ITD+ AML patient samples were collected after informed consent. Mononuclear blasts were isolated through Ficoll (Axis-Shield) density centrifugation, and cell viability was assessed by a Trypan Blue Exclusion Assay. Protocols for sample handling and data analysis were approved by the Tongji Hospital Ethics Committee and were performed in compliance with the Declaration of Helsinki. Patient information is shown in supplemental Table 1 (available on the Blood Web site).
Mice
NOD-SCID mice (Charles River) were used for the MV4-11 xenograft model, and NOG mice (Charles River) were used for the human AML PDX model. All experiments were carried out according to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. Details are provided in supplemental Materials.
Statistics
Data visualizing and statistical analysis were performed using GraphPad Prism software. Survival comparisons were performed using the Kaplan-Meier/log-rank test. Other differences between experimental groups were analyzed using a paired or unpaired Student t test and 2-way analysis of variance with multiple testing. A P value < .05 was considered significant in all experiments.
Results
HDAC8 is upregulated upon FLT3 inhibition
We previously discovered that common chemotherapy reagents doxorubicin and cytarabine could upregulate HDAC3 in AML cells.19 We confirmed this phenomenon in 2 other AML cell lines: MV4-11 and MOLM-13 (supplemental Figure 1A-B). Because MV4-11 and MOLM-13 cells contain FLT3-ITD mutation, and FLT3 inhibitors are used as monotherapy or in combination with chemotherapy to treat FLT3-ITD+ AML in clinical practice, we further evaluated the expression of class I HDACs in both cell lines following FLT3 inhibitor AC220 (quizartinib) treatment. Interestingly, HDAC8 was upregulated upon AC220 exposure, whereas other class I members did not show any change (supplemental Figure 1C-D). To understand comprehensively about the possible roles of HDACs in FLT3-ITD+ AML pathogenesis and drug resistance, we next examined the transcriptional alterations of 11 HDACs in both cells lines following AC220 treatment and found that HDAC8 was the most significantly upregulated gene (Figure 1A-B). Treating MV4-11 and MOLM-13 cells with different concentrations of AC220 revealed a dose-dependent escalation of HDAC8 at the messenger RNA (mRNA) and protein levels (Figure 1C-F). AC220 treatment also increased the transcription of HDAC8 in a time-dependent manner (supplemental Figure 1E-F). Downregulation of FLT3 activity using 2 other inhibitors, PKC412 (midostaurin) and PRT062607 (a spleen TKI known to reduce FLT3 downstream signaling23 ), as well as small interfering RNA (siRNA)-mediated knockdown of FLT3, also resulted in increased expression of HDAC8, validating that upregulation of HDAC8 was indeed due to FLT3 signaling inhibition rather than off-target effects (Figure 1G-J; supplemental Figure 1G-H). Treatment of primary FLT3-ITD+ AML samples with AC220 also confirmed HDAC8 upregulation (Figure 1K-L).
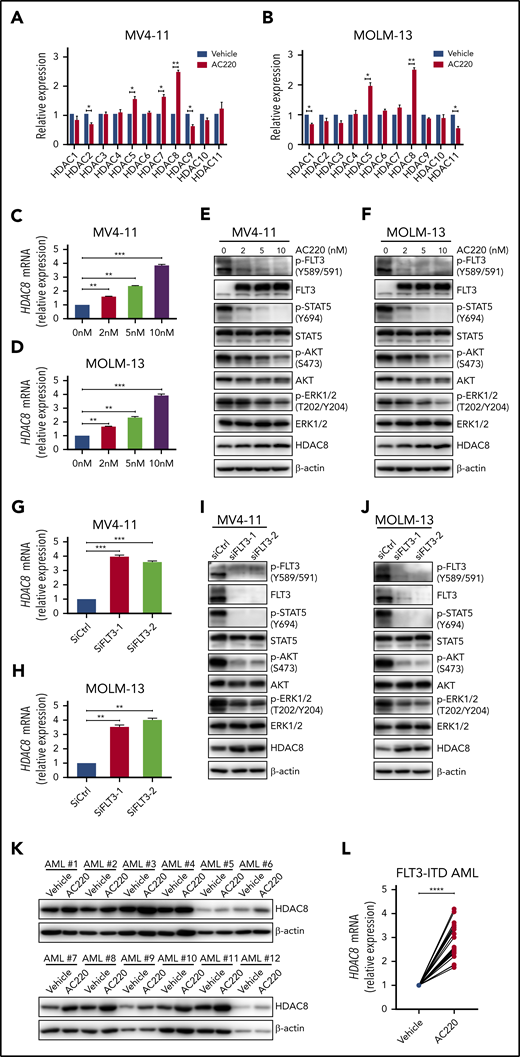
FLT3 inhibition induces HDAC8 upregulation. MV4-11 cells (A) and MOLM-13 cells (B) were treated with AC220 (5 nM) for 24 hours. The transcription of 11 HDACs was analyzed by quantitative real-time polymerase chain reaction (PCR). MV4-11 cells (C) and MOLM-13 cells (D) were treated with different concentrations of AC220 for 24 hours. The transcription of HDAC8 was analyzed by quantitative real-time PCR. MV4-11 cells (E) and MOLM-13 cells (F) were treated with different concentrations of AC220 for 24 hours and then subjected to western blot to detect the indicated proteins. MV4-11 cells (G) and MOLM-13 cells (H) cells were transfected with FLT3 siRNA (siFLT3-1 and siFLT3-2) and control siRNA (siCtrl). The transcription of HDAC8 was analyzed by quantitative real-time PCR. MV4-11 cells (I) and MOLM-13 cells (J) were transfected with FLT3 siRNA and control siRNA and then subjected to western blot to detect the indicated proteins. (K) Primary AML blasts from 12 FLT3-ITD+ AML patients were treated with AC220 (20 nM) for 24 hours and then analyzed by immunoblot for HDAC8. (L) Primary AML blasts from 12 FLT3-ITD+ AML patients were treated with AC220 (20 nM) for 24 hours. The transcription of HDAC8 was analyzed by quantitative real-time PCR. Data are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
FLT3 inhibition induces HDAC8 upregulation. MV4-11 cells (A) and MOLM-13 cells (B) were treated with AC220 (5 nM) for 24 hours. The transcription of 11 HDACs was analyzed by quantitative real-time polymerase chain reaction (PCR). MV4-11 cells (C) and MOLM-13 cells (D) were treated with different concentrations of AC220 for 24 hours. The transcription of HDAC8 was analyzed by quantitative real-time PCR. MV4-11 cells (E) and MOLM-13 cells (F) were treated with different concentrations of AC220 for 24 hours and then subjected to western blot to detect the indicated proteins. MV4-11 cells (G) and MOLM-13 cells (H) cells were transfected with FLT3 siRNA (siFLT3-1 and siFLT3-2) and control siRNA (siCtrl). The transcription of HDAC8 was analyzed by quantitative real-time PCR. MV4-11 cells (I) and MOLM-13 cells (J) were transfected with FLT3 siRNA and control siRNA and then subjected to western blot to detect the indicated proteins. (K) Primary AML blasts from 12 FLT3-ITD+ AML patients were treated with AC220 (20 nM) for 24 hours and then analyzed by immunoblot for HDAC8. (L) Primary AML blasts from 12 FLT3-ITD+ AML patients were treated with AC220 (20 nM) for 24 hours. The transcription of HDAC8 was analyzed by quantitative real-time PCR. Data are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Targeting HDAC8 enhances AC220-mediated inhibition of FLT3-ITD+ AML
Because deregulated HDAC8 expression has been implicated in promoting tumor proliferation and drug resistance in a variety of cancers,24 we reasoned that HDAC8 upregulation in FLT3-ITD+ AML cells upon FLT3 inhibition might serve as an important mechanism to promote the survival of leukemia cells and offset the leukemia-killing effect of TKIs. We first downregulated HDAC8 expression in MV4-11 and MOLM-13 cells through lentivirus vectors expressing HDAC8 short hairpin RNA (shRNA; shHDAC8) (supplemental Figure 2A). HDAC8 knockdown readily caused significant apoptosis of both cell lines, and the antileukemia effect was more pronounced when combined with AC220 (Figure 2A). Pharmacological inhibition of HDAC8 activity with an HDAC8-selective inhibitor 22d14,25 significantly enhanced AC220-induced apoptosis and growth inhibition of MV4-11 and MOLM-13 cells (Figure 2B-C). 22d also synergized with AC220 and cytarabine to eliminate FLT3-ITD+ AML cells (supplemental Figure 2B). Notably, 22d treatment had no effect on myeloid differentiation (supplemental Figure 2C). Consistent with previous reports,21 AC220 modestly inhibited the survival of primary human FLT3-ITD+ AML cells (Figure 2D-E), whereas the combination of 22d plus AC220 significantly reduced the survival and colony-forming capacity of FLT3-ITD+ AML cells compared with AC220 or 22d alone (Figure 2D-F). shHDAC8 plus AC220 also enhanced suppression of FLT3-ITD+ AML cell survival and colony formation compared with AC220 alone (Figure 2G-H). Finally, we investigated the effect of HDAC8 inhibition in vivo using xenotransplantation experiments. Inhibiting HDAC8 together with FLT3 resulted in a much greater antileukemia effect than downregulating HDAC8 or FLT3 alone, as demonstrated by the significantly prolonged survival of leukemia mice (Figure 2I-J). Taken together, these findings indicate that HDAC8 inhibition enhances targeting of FLT3-ITD+ AML in combination with TKIs.
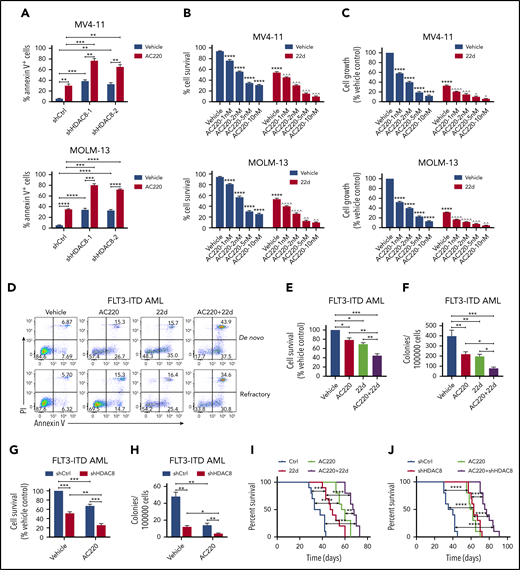
HDAC8 inhibition enhances AC220-mediated elimination of FLT3-ITD+ AML. (A) MV4-11 cells and MOLM-13 cells were transduced with lentiviral vectors expressing doxycycline-inducible control shRNA (shCtrl) or HDAC8 shRNA (shHDAC8-1 and shHDAC8-2). Doxycycline was added to induce shRNA expression for 48 hours. Then cells were treated with vehicle (dimethyl sulfoxide) or AC220 (5 nM) for another 24 hours. The apoptosis of cells was analyzed by annexin V/propidium iodide (PI) labeling. (B-C) MV4-11 and MOLM-13 cells were treated with the indicated concentrations of AC220 and/or 22d (10 μM). (B) The survival of cells was analyzed by annexin V/PI labeling. (C) Cell growth was evaluated using a Cell Counting Kit-8 (CCK-8) assay. ****P < .0001 vs vehicle control; ^P < .05, ^^P < .01, ^^^P < .001, ^^^^P < .0001 vs AC220 treatment alone. (D) Primary FLT3-ITD+ AML blasts were treated as indicated (AC220, 20 nM; 22d, 10 μM), and apoptosis was analyzed by annexin V/PI labeling. (E) Primary FLT3-ITD+ AML (n = 6) blasts were treated with AC220 (20 nM), 22d (10 μM), or the combination for 48 hours. Cell survival was analyzed by annexin V/PI labeling. (F) FLT3-ITD+ AML (n = 6) CD34+ cells, treated as indicated, were plated in methylcellulose culture, and erythroid or myeloid colonies were enumerated after 14 days. (G) Primary FLT3-ITD+ AML (n = 4) blasts, transduced with shCtrl or shHDAC8 vectors, were cultured or not with AC220 (20 nM) for 72 hours. Cell survival was analyzed by annexin V/PI labeling. (H) FLT3-ITD+ AML (n = 4) CD34+ cells, transduced with shCtrl or shHDAC8 vectors, were cultured or not with AC220 (20 nM) for 72 hours. Colony-formation assays were performed, and erythroid or myeloid colonies were enumerated after 14 days. (I) NOD-SCID mice were transplanted with MV4-11 cells (2 × 106 per mouse) and treated as indicated (AC220, 10 mg/kg per day; 22d, 100 mg/kg per day). The survival of diseased mice was monitored. (J) MV4-11 cells transduced with shCtrl or shHDAC8 vectors were transplanted into NOD-SCID mice, which were then treated as indicated. The survival of diseased mice was monitored. Data are mean ± standard error of the mean. In (A) and (E-J), *P < .05, **P < .01, ***P < .001, ****P < .0001.
HDAC8 inhibition enhances AC220-mediated elimination of FLT3-ITD+ AML. (A) MV4-11 cells and MOLM-13 cells were transduced with lentiviral vectors expressing doxycycline-inducible control shRNA (shCtrl) or HDAC8 shRNA (shHDAC8-1 and shHDAC8-2). Doxycycline was added to induce shRNA expression for 48 hours. Then cells were treated with vehicle (dimethyl sulfoxide) or AC220 (5 nM) for another 24 hours. The apoptosis of cells was analyzed by annexin V/propidium iodide (PI) labeling. (B-C) MV4-11 and MOLM-13 cells were treated with the indicated concentrations of AC220 and/or 22d (10 μM). (B) The survival of cells was analyzed by annexin V/PI labeling. (C) Cell growth was evaluated using a Cell Counting Kit-8 (CCK-8) assay. ****P < .0001 vs vehicle control; ^P < .05, ^^P < .01, ^^^P < .001, ^^^^P < .0001 vs AC220 treatment alone. (D) Primary FLT3-ITD+ AML blasts were treated as indicated (AC220, 20 nM; 22d, 10 μM), and apoptosis was analyzed by annexin V/PI labeling. (E) Primary FLT3-ITD+ AML (n = 6) blasts were treated with AC220 (20 nM), 22d (10 μM), or the combination for 48 hours. Cell survival was analyzed by annexin V/PI labeling. (F) FLT3-ITD+ AML (n = 6) CD34+ cells, treated as indicated, were plated in methylcellulose culture, and erythroid or myeloid colonies were enumerated after 14 days. (G) Primary FLT3-ITD+ AML (n = 4) blasts, transduced with shCtrl or shHDAC8 vectors, were cultured or not with AC220 (20 nM) for 72 hours. Cell survival was analyzed by annexin V/PI labeling. (H) FLT3-ITD+ AML (n = 4) CD34+ cells, transduced with shCtrl or shHDAC8 vectors, were cultured or not with AC220 (20 nM) for 72 hours. Colony-formation assays were performed, and erythroid or myeloid colonies were enumerated after 14 days. (I) NOD-SCID mice were transplanted with MV4-11 cells (2 × 106 per mouse) and treated as indicated (AC220, 10 mg/kg per day; 22d, 100 mg/kg per day). The survival of diseased mice was monitored. (J) MV4-11 cells transduced with shCtrl or shHDAC8 vectors were transplanted into NOD-SCID mice, which were then treated as indicated. The survival of diseased mice was monitored. Data are mean ± standard error of the mean. In (A) and (E-J), *P < .05, **P < .01, ***P < .001, ****P < .0001.
HDAC8 attenuates p53 activation to promote FLT3-ITD+ AML survival and TKI resistance
To explore the mechanism of HDAC8 that promotes FLT3-ITD+ AML survival and TKI resistance, we used RNA sequencing to investigate transcriptomic changes in MV4-11 cells upon 22d treatment. The differentially expressed genes predicted activated cell death, upregulated DNA damage, reduced cell survival and cell proliferation, and downregulated homologous recombination in 22d-treated MV4-11 cells (Figure 3A). Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that the p53 signaling pathway was significantly upregulated (Figure 3B). Upstream regulator prediction by Ingenuity Pathway Analysis also showed p53 to be the key activated upstream transcription factor (Figure 3C). Indeed, p53 target genes were significantly upregulated following 22d treatment (supplemental Figure 3A). Therefore, we reasoned that HDAC8 might regulate FLT3-ITD+ AML maintenance upon TKI exposure through modulating the activity of p53. MOLM-13 and MV4-11 cells (p53 wild-type [WT] and FLT3-ITD) showed significantly higher sensitivity to 22d compared with OCI-AML3 cells (p53 WT and FLT3-WT), NB4 cells (p53 mutant and FLT3-WT), and HL-60, THP-1, and U937 cells (p53 null and FLT3 WT) (supplemental Figure 3B), indicating that the sensitivity of 22d was associated with intracellular p53 and FLT3 status. 22d pretreatment significantly increased the expression of p53 target genes in MV4-11 and MOLM-13 cells following irradiation (supplemental Figure 3C), whereas knockdown of p53 reduced the sensitivity of both cell lines to 22d and inhibited the apoptosis induced by irradiation and 22d (Figure 3D; supplemental Figure 3D-E).
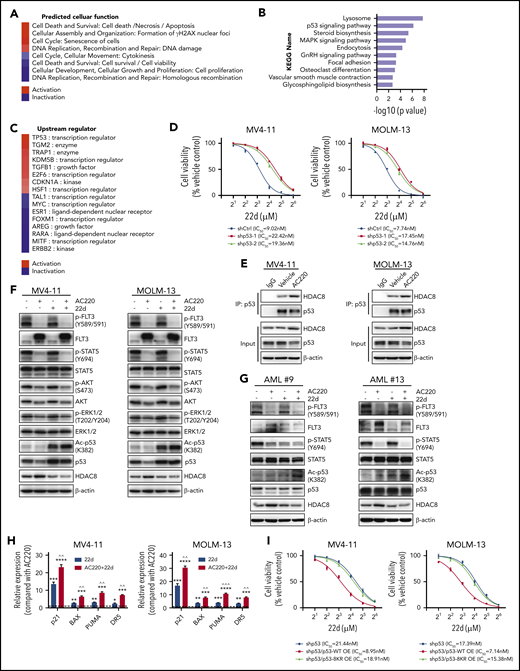
HDAC8 inhibits p53 activity to promote leukemia maintenance and TKI resistance. (A-C) MV4-11 cells were treated with dimethyl sulfoxide or 22d (10 μM) for 18 hours and then subjected to RNA-sequencing analysis. Differentially expressed genes were identified (P < .001). (A) Cellular functions were predicted on the basis of the overlap of differentially expressed genes with Ingenuity Pathway Analysis (IPA), ranked based on the calculated Z score. Red represents predicted activation, and blue represents predicted inhibition of the respective cellular function category in 22d vs vehicle condition. (B) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of differentially expressed genes. (C) Activation and inhibition of upstream regulators predicted with IPA. The upstream regulators were ranked based on Z score. Red represents predicted activation, and blue represents predicted inhibition of the upstream regulator. (D) p53 shRNA (shp53-1 and shp53-2) or control shRNA (shCtrl) was expressed in MV4-11 and MOLM-13 cells. Viability of cells exposed to different concentrations of 22d for 48 hours was analyzed by a Cell Counting Kit-8 (CCK-8) assay. (E) MV4-11 and MOLM-13 cells were treated with vehicle or AC220 (5 nM) for 24 hours. Protein lysates were immunoprecipitated with p53 antibody and then immunoblotted for HDAC8 and p53. (F) MV4-11 and MOLM-13 cells were treated with AC220 (5 nM), 22d (10 μM), or both for 24 hours and then subjected to western blot to detect the indicated proteins. (G) Primary FLT3-ITD+ AML blasts were treated with AC220 (20 nM), 22d (10 μM), or both for 24 hours and then subjected to western blot to detect the indicated proteins. (H) MV4-11 and MOLM-13 cells were treated with AC220 (5 nM), 22d (10 μM), or both for 24 hours. The relative expression of the indicated p53 target genes was analyzed and compared with AC220 treatment alone. (I) MV4-11 and MOLM-13 cells with p53 knockdown were transduced with vectors expressing WT p53 (p53-WT) or acetylation defect mutant p53 (a p53 mutant with 8 potential acetylation sites mutated; p53-8KR). Viability of cells exposed to 22d for 48 hours was analyzed using a CCK-8 assay. Data are mean ± standard error of the mean. **P < .01, ***P < .001, ****P < .0001 vs AC220; ^^P < .01, ^^^P < .001 vs 22d. IC50, half maximum inhibitory concentraton; OE, overexpression.
HDAC8 inhibits p53 activity to promote leukemia maintenance and TKI resistance. (A-C) MV4-11 cells were treated with dimethyl sulfoxide or 22d (10 μM) for 18 hours and then subjected to RNA-sequencing analysis. Differentially expressed genes were identified (P < .001). (A) Cellular functions were predicted on the basis of the overlap of differentially expressed genes with Ingenuity Pathway Analysis (IPA), ranked based on the calculated Z score. Red represents predicted activation, and blue represents predicted inhibition of the respective cellular function category in 22d vs vehicle condition. (B) Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of differentially expressed genes. (C) Activation and inhibition of upstream regulators predicted with IPA. The upstream regulators were ranked based on Z score. Red represents predicted activation, and blue represents predicted inhibition of the upstream regulator. (D) p53 shRNA (shp53-1 and shp53-2) or control shRNA (shCtrl) was expressed in MV4-11 and MOLM-13 cells. Viability of cells exposed to different concentrations of 22d for 48 hours was analyzed by a Cell Counting Kit-8 (CCK-8) assay. (E) MV4-11 and MOLM-13 cells were treated with vehicle or AC220 (5 nM) for 24 hours. Protein lysates were immunoprecipitated with p53 antibody and then immunoblotted for HDAC8 and p53. (F) MV4-11 and MOLM-13 cells were treated with AC220 (5 nM), 22d (10 μM), or both for 24 hours and then subjected to western blot to detect the indicated proteins. (G) Primary FLT3-ITD+ AML blasts were treated with AC220 (20 nM), 22d (10 μM), or both for 24 hours and then subjected to western blot to detect the indicated proteins. (H) MV4-11 and MOLM-13 cells were treated with AC220 (5 nM), 22d (10 μM), or both for 24 hours. The relative expression of the indicated p53 target genes was analyzed and compared with AC220 treatment alone. (I) MV4-11 and MOLM-13 cells with p53 knockdown were transduced with vectors expressing WT p53 (p53-WT) or acetylation defect mutant p53 (a p53 mutant with 8 potential acetylation sites mutated; p53-8KR). Viability of cells exposed to 22d for 48 hours was analyzed using a CCK-8 assay. Data are mean ± standard error of the mean. **P < .01, ***P < .001, ****P < .0001 vs AC220; ^^P < .01, ^^^P < .001 vs 22d. IC50, half maximum inhibitory concentraton; OE, overexpression.
Because previous studies have demonstrated that HDAC8 could bind to and deacetylate p53,14,26 we performed a coimmunoprecipitation assay to analyze the association between p53 and HDAC8 in MV4-11 and MOLM-13 cells. Western blot showed that HDAC8 did interact with p53, and their association was enhanced under AC220 treatment conditions (Figure 3E). Targeting FLT3 in MV4-11 and MOLM-13 cells had little effect on the acetylation of p53 (Figure 3F-G; supplemental Figure 3F), indicating that HDAC8 upregulation might serve as a feedback mechanism in FLT3-ITD+ AML to deacetylate p53, downregulate its activity (supplemental Figure 3F-G), and, thus, maintain leukemia survival under TKI treatment. Indeed, inhibiting HDAC8 by 22d or shRNA-mediated knockdown markedly increased p53 acetylation and activation, and this effect was even more significant when FLT3 and HDAC8 activities were suppressed simultaneously (Figure 3F-H; supplemental Figure 3F). Depletion of p53 partially rescued FLT3-ITD+ AML cells from 22d- and/or AC220-induced apoptosis (supplemental Figure 3H), whereas ectopically expressing WT p53, but not acetylation defect mutant p53 (a p53 mutant with 8 potential acetylation sites mutated, p53-8KR), in p53-depleted cells resensitized these cells to 22d (Figure 3I). These data support that HDAC8 promotes the maintenance of FLT3-ITD+ AML upon TKI treatment.
FOXO induces HDAC8 transcription following TKI treatment
We next studied the mechanism underlying HDAC8 upregulation upon FLT3 inhibition. Treating MV4-11 and MOLM-13 cells with actinomycin D or cycloheximide abrogated HDAC8 upregulation following AC220 exposure (supplemental Figure 4A-B), indicating that HDAC8 upregulation was due to increased transcription. We then looked for transcriptional binding motifs throughout the promoter region of HDAC8 and found that there were cis elements potentially bound by FOXO1 and FOXO3 located within 400 base pairs upstream of the transcription start site of HDAC8 (supplemental Figure 4C). FOXO1 and FOXO3 are downstream targets of AKT and ERK, which could phosphorylate FOXO1 and FOXO3, reduce the transcriptional activity of both factors, and promote their degradation. Treating MV4-11 and MOLM-13 cells with AC220 inhibited AKT and ERK activity, as well as increased the protein levels of FOXO1 and FOXO3 (Figure 4A-B). Inhibition of FLT3 downstream signaling pathways AKT, ERK1/2, and STAT5, individually or in combination, revealed that upregulation of HDAC8, increased expression of FOXO1 and FOXO3, and downregulated activity of AKT and ERK1/2 were closely correlated (Figure 4A-B). Knockdown of FOXO1 or FOXO3 alone reduced the inducing effect of AC220 on HDAC8 transcription only slightly (supplemental Figure 4D). We reasoned that FOXO1 and FOXO3 might participate in HDAC8 transcription collaboratively, and elevated expression of 1 factor following FLT3 inhibition would offset the depletion effect of the other factor. Luciferase-reporter assay showed that FOXO1 or FOXO3 alone readily induced HDAC8 transcription, and the inducing effect was more significant when both factors were coexpressed (supplemental Figure 4E). Deletion of a FOXO1 and/or a FOXO3 consensus binding motif diminished the transcription activating effect (supplemental Figure 4E). Indeed, depletion of FOXO1 and FOXO3 simultaneously in MV4-11 and MOLM-13 cells abrogated HDAC8 upregulation at the mRNA and protein levels under AC220 treatment (Figure 4C-D; supplemental Figure 4F), whereas expressing FOXO1 and FOXO3 increased the transcription of HDAC8 (Figure 4E-F; supplemental Figure 4G). Electrophoretic mobility shift assay demonstrated that FOXO1 and FOXO3 directly bound to the HDAC8 promoter but not to the binding motif–mutated DNA probes (supplemental Figure 4H). Moreover, AC220 treatment promoted the binding of FOXO1 and FOXO3 to HDAC8 promoter (Figure 4G-H).
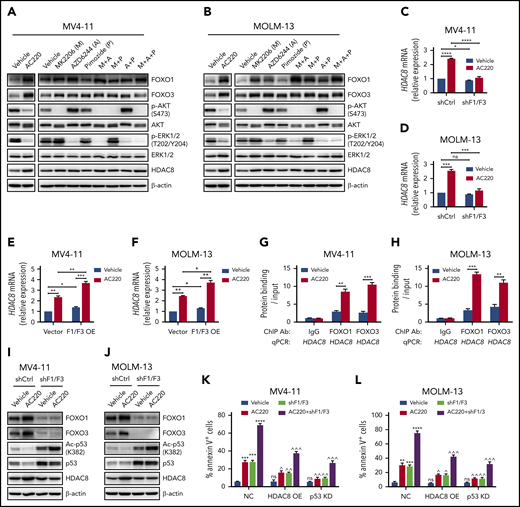
FOXOs induce HDAC8 transcription upon FLT3 inhibitor treatment. MV4-11 (A) and MOLM-13 (B) cells were treated with different inhibitors individually or in combination, as indicated, for 24 hours. Expression of the indicated proteins was analyzed with western blot. MV4-11 (C) and MOLM-13 (D) cells were transduced with vectors expressing control shRNA (shCtrl) or FOXO1 shRNA plus FOXO3 shRNA (shF1/F3). Doxycycline was added to induce shRNA expression for 48 hours. Then cells were treated with vehicle (dimethyl sulfoxide) or AC220 (5 nM) for another 24 hours. HDAC8 transcription was analyzed with quantitative real-time polymerase chain reaction (PCR). ns, P > .05, *P < .05, *** P < .001, ****P < .0001. MV4-11 (E) and MOLM-13 (F) cells, transduced with empty vectors (Vector) or FOXO1 plus FOXO3 expressing vectors (F1/F3 OE), were treated with AC220 (5 nM) for 24 hours. HDAC8 transcription was analyzed with quantitative real-time PCR. *P < .05, **P < .01, ***P < .001. Chromatin immunoprecipitation–quantitative PCR analysis of FOXO1 and FOXO3 binding to HDAC8 promoter in MV4-11 (G) and MOLM-13 (H) cells treated with vehicle or AC220 (5 nM). **P < .01, ***P < .001. MV4-11 (I) and MOLM-13 (J) cells, transduced with shCtrl or shF1/F3, were treated with vehicle or AC220 (5 nM) for 24 hours and subjected to western blot to detect the indicated proteins. shCtrl or shF1/F3 transduced MV4-11 (K) and MOLM-13 (L) cells (NC) were transduced with vectors expressing HDAC8 (HDAC8 OE) or p53 shRNA (p53 KD). Doxycycline was added to induce shRNA expression for 48 hours and then cells were treated with vehicle (dimethyl sulfoxide) or AC220 (5 nM) for another 24 hours. The apoptosis of cells was analyzed with annexin V/propidium iodide labeling. **P < .01, ***P < .001, ****P < .0001 vs vehicle; ^P < .05, ^^P < .01, ^^^P < .001 vs NC. Data are mean ± standard error of the mean. ns, not significant (P > .05). KD, knockdown; NC, negative control; OE, overexpression.
FOXOs induce HDAC8 transcription upon FLT3 inhibitor treatment. MV4-11 (A) and MOLM-13 (B) cells were treated with different inhibitors individually or in combination, as indicated, for 24 hours. Expression of the indicated proteins was analyzed with western blot. MV4-11 (C) and MOLM-13 (D) cells were transduced with vectors expressing control shRNA (shCtrl) or FOXO1 shRNA plus FOXO3 shRNA (shF1/F3). Doxycycline was added to induce shRNA expression for 48 hours. Then cells were treated with vehicle (dimethyl sulfoxide) or AC220 (5 nM) for another 24 hours. HDAC8 transcription was analyzed with quantitative real-time polymerase chain reaction (PCR). ns, P > .05, *P < .05, *** P < .001, ****P < .0001. MV4-11 (E) and MOLM-13 (F) cells, transduced with empty vectors (Vector) or FOXO1 plus FOXO3 expressing vectors (F1/F3 OE), were treated with AC220 (5 nM) for 24 hours. HDAC8 transcription was analyzed with quantitative real-time PCR. *P < .05, **P < .01, ***P < .001. Chromatin immunoprecipitation–quantitative PCR analysis of FOXO1 and FOXO3 binding to HDAC8 promoter in MV4-11 (G) and MOLM-13 (H) cells treated with vehicle or AC220 (5 nM). **P < .01, ***P < .001. MV4-11 (I) and MOLM-13 (J) cells, transduced with shCtrl or shF1/F3, were treated with vehicle or AC220 (5 nM) for 24 hours and subjected to western blot to detect the indicated proteins. shCtrl or shF1/F3 transduced MV4-11 (K) and MOLM-13 (L) cells (NC) were transduced with vectors expressing HDAC8 (HDAC8 OE) or p53 shRNA (p53 KD). Doxycycline was added to induce shRNA expression for 48 hours and then cells were treated with vehicle (dimethyl sulfoxide) or AC220 (5 nM) for another 24 hours. The apoptosis of cells was analyzed with annexin V/propidium iodide labeling. **P < .01, ***P < .001, ****P < .0001 vs vehicle; ^P < .05, ^^P < .01, ^^^P < .001 vs NC. Data are mean ± standard error of the mean. ns, not significant (P > .05). KD, knockdown; NC, negative control; OE, overexpression.
We then investigated the effect of FOXO1 and FOXO3 codepletion on HDAC8 downstream signaling. Knockdown of both factors modestly increased the acetylation of p53, as well as caused apoptosis and growth inhibition in FLT3-ITD+ AML cells (Figure 4I-J; supplemental Figure 4I-J). AC220 treatment plus FOXO1/3 suppression further increased p53 acetylation (Figure 4I-J) and resulted in significantly greater apoptosis (Figure 4K-L; supplemental Figure 4I), which could be partially rescued by HDAC8 overexpression or p53 depletion (Figure 4K-L). Taken together, these results suggest that FOXO1 and FOXO3 induce HDAC8 transcription to promote TKI resistance.
HDAC8 targeting enhances elimination of FLT3-ITD+ AML in vivo in combination with TKIs and reduces the leukemia-initiating capacity
To translate our experimental results into clinical usage, we tested the in vivo effect of 22d, AC220, or both on the PDX model engrafted with primary human FLT3-ITD+ AML samples (Figure 5A). Following confirmation of human leukemia engraftment in murine peripheral blood (>5% human CD45+ cells), mice were treated with vehicle (control), AC220 (10 mg/kg per day, gavage), 22d (100 mg/kg per day, intraperitoneal), or the combination for 4 weeks. Engrafted leukemia cells were human CD45+ and CD33+ and expressed the FLT3-ITD gene (data not shown). AC220 or 22d treatment reduced the percentage and total number of human CD45+ leukemia cells (Figure 5B-D; supplemental Figure 5A-B) and CD45+CD34+ leukemia cells (Figure 5E-F; supplemental Figure 5C-D) in murine bone marrow, with a further reduction seen in mice treated with AC220 plus 22d (Figure 5B-F; supplemental Figure 5A-D). Notably, mice treated with AC220 or 22d alone exhibited improved survival after discontinuation of treatment compared with control mice, and the combination of AC220 and 22d further prolonged the survival period (Figure 5G). Intriguingly, when evaluating the immunophenotype of the remaining human CD45+ cells in murine bone marrow, we found that the frequency of CD34+ cells within human CD45+ cells was much higher in AC220-treated mice than in control mice, whereas 22d treatment significantly reduced the percentage of CD34+ cell within the remaining human CD45+ cells (Figure 5H; supplemental Figure 5E-F). These results indicate that TKIs cannot eliminate FLT3-ITD+ AML stem cells, but targeting HDAC8 can eradicate these cells. Consistent with these findings, a leukemia-initiating capacity assay by secondary transplantation revealed that the AML burden in mice receiving AC220-treated leukemia cells was comparable to or higher than that in mice receiving control leukemia cells (Figure 5G-H). In contrast, secondary transplantation of leukemia cells from mice receiving 22d treatment or combination treatment resulted in significantly lower leukemia burden, indicating the reduced leukemia-initiating capacity of residual cells (Figure 5G-H; supplemental Figure 5G-H). Taken together, these results show that the combination targeting FLT3 and HDAC8 enhances elimination of FLT3-ITD+ AML in vivo, and HDAC8 inhibition reduces the leukemia-initiating capacity.
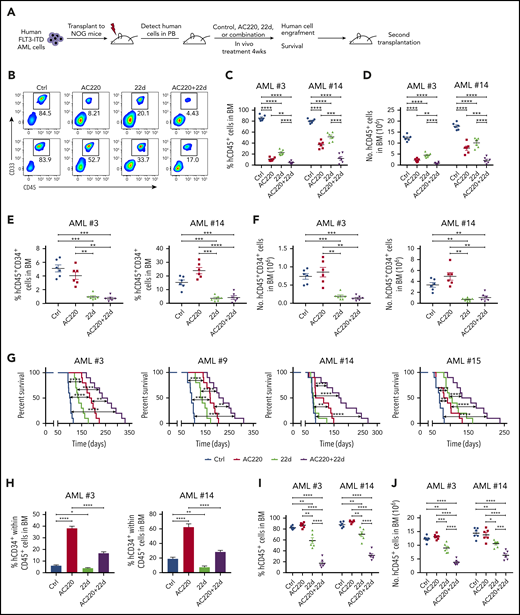
HDAC8 inhibition enhances elimination of FLT3-ITD+ AML in vivo in combination with TKIs. (A) T-cell–depleted primary human FLT3-ITD+ AML cells were injected into sublethally irradiated (250 cGy) NOG mice (2 × 106 cells per mouse). After engraftment was confirmed, mice were treated for 4 weeks with vehicle (control), AC220 (10 mg/kg per day, by mouth), 22d (100 mg/kg per day, intraperitoneally), or AC220 + 22d (n = 20 per group). Engraftment of human cells was analyzed by flow cytometry. Survival analysis and secondary transplantation were also performed. (B) Representative results for CD45 and CD33 expression from AML #3 and AML #14. Percentage (C) and number (D) of human CD45+ cells from AML #3 and AML #14 in the bone marrow (BM) of treated mice. Percentage (E) and number (F) of human CD45+CD34+ cells from AML #3 and AML #14 in the BM of treated mice. (G) The survival of mice transplanted with 4 different samples was displayed respectively (n = 10 per group). (H) Percentage of CD34+ cells within the human CD45+ population from AML #3 and AML #14. Percentage (I) and number (J) of human CD45+ cells (AML #3 and AML #14) in the BM of secondary recipient mice at 12 weeks. Data are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
HDAC8 inhibition enhances elimination of FLT3-ITD+ AML in vivo in combination with TKIs. (A) T-cell–depleted primary human FLT3-ITD+ AML cells were injected into sublethally irradiated (250 cGy) NOG mice (2 × 106 cells per mouse). After engraftment was confirmed, mice were treated for 4 weeks with vehicle (control), AC220 (10 mg/kg per day, by mouth), 22d (100 mg/kg per day, intraperitoneally), or AC220 + 22d (n = 20 per group). Engraftment of human cells was analyzed by flow cytometry. Survival analysis and secondary transplantation were also performed. (B) Representative results for CD45 and CD33 expression from AML #3 and AML #14. Percentage (C) and number (D) of human CD45+ cells from AML #3 and AML #14 in the bone marrow (BM) of treated mice. Percentage (E) and number (F) of human CD45+CD34+ cells from AML #3 and AML #14 in the BM of treated mice. (G) The survival of mice transplanted with 4 different samples was displayed respectively (n = 10 per group). (H) Percentage of CD34+ cells within the human CD45+ population from AML #3 and AML #14. Percentage (I) and number (J) of human CD45+ cells (AML #3 and AML #14) in the BM of secondary recipient mice at 12 weeks. Data are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Effects of combined HDAC8 inhibition and TKIs extend to other tyrosine kinase–activating mutations
Next, to investigate whether a similar HDAC8 upregulation phenomenon occurs in leukemia with other activated tyrosine kinases upon TKI treatment, we analyzed the consequences of inhibiting KIT tyrosine mutation, which is frequently detected in core-binding factor AML and confers an unfavorable prognosis, using dasatinib. In a KITN822K-positive cell line, Kasumi-1, dasatinib treatment resulted in upregulation of HDAC8 at the mRNA and protein levels (supplemental Figure 6A-B). We then used a murine leukemia model driven by AML1-ETO and KITN822K coexpression to determine whether combined targeting of tyrosine kinase and HDAC8 would also show a superior antileukemia effect. Indeed, dasatinib plus 22d treatment significantly prolonged the survival of leukemia mice compared with dasatinib or 22d treatment alone (supplemental Figure 6C). Also, compared with a single agent, combination treatment significantly reduced the leukemia burden in peripheral blood, bone marrow, and spleen (supplemental Figure 6D-H).
Discussion
Despite the recent advances in AML therapy, the 5-year survival rate of FLT3-ITD+ AML patients remains very low.27 Although several small molecule FLT3 TKIs have been developed for FLT3-ITD+ AML treatment, these TKIs have shown only modest responses in clinical settings as monotherapy, and the majority of FLT3-ITD+ AML patients would relapse very shortly.4,5 Therefore, it is pressing to elucidate the mechanisms underlying TKI resistance and develop new strategies to eliminate FLT3-ITD+ AML more effectively. Here, we demonstrated that HDAC8 was upregulated upon FLT3 inhibition. Targeting HDAC8 enhanced TKI-mediated killing of FLT3-ITD+ AML cells. The effect of HDAC8 inhibition on FLT3-ITD+ AML cells was related to increased p53 acetylation activity. These results indicate a critical role for HDAC8-mediated downregulation of p53 activity in maintaining FLT3-ITD+ AML under TKI treatment and promoting drug resistance (Figure 6). Importantly, using PDX models, we demonstrated that the combination of HDAC8 inhibition plus FLT3 TKI AC220 significantly enhanced elimination of FLT3-ITD+ AML in vivo and reduced the frequency of CD34+ cells within human CD45+ leukemia cells compared with AC220 alone, suggesting that HDAC8 suppression may successfully reduce the leukemia-initiating capacity.
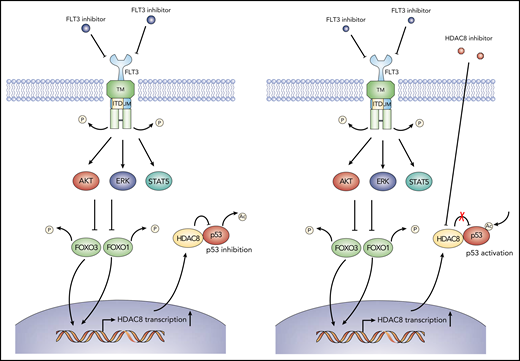
Schematic model showing the action of mechanism of combined FLT3 and HDAC8 inhibition. When FLT3-ITD+ AML cells are exposed to FLT3 inhibitors, FOXO1 and FOXO3 are activated following the downregulation of FLT3 signaling and downstream AKT and ERK signaling (left panel). Activated FOXO1 and FOXO3 bind to HDAC8 promoter and induce its transcription, which, in turn, associates with and inhibits the antitumor activity of p53 via deacetylation, thus promoting the maintenance and TKI resistance of FLT3-ITD+ AML. Combining HDAC8 inhibitors with FLT3 inhibitors abrogates the inhibitory effect of HDAC8 on p53 and increases p53 acetylation, thus significantly enhancing the elimination of FLT3-ITD+ AML (right panel).
Schematic model showing the action of mechanism of combined FLT3 and HDAC8 inhibition. When FLT3-ITD+ AML cells are exposed to FLT3 inhibitors, FOXO1 and FOXO3 are activated following the downregulation of FLT3 signaling and downstream AKT and ERK signaling (left panel). Activated FOXO1 and FOXO3 bind to HDAC8 promoter and induce its transcription, which, in turn, associates with and inhibits the antitumor activity of p53 via deacetylation, thus promoting the maintenance and TKI resistance of FLT3-ITD+ AML. Combining HDAC8 inhibitors with FLT3 inhibitors abrogates the inhibitory effect of HDAC8 on p53 and increases p53 acetylation, thus significantly enhancing the elimination of FLT3-ITD+ AML (right panel).
HDAC8 is expressed in a variety of cancers, including leukemia, and shows a tendency toward higher expression in cancer tissues.28,29 Several studies have demonstrated the importance of HDAC in leukemia maintenance and drug resistance. In adult T-cell leukemia/lymphoma, HDAC8 inhibition suppresses cell growth and induces calcium-mediated and caspase-dependent apoptosis.30,31 AML-associated inv(16) fusion protein binds HDAC8 and p53 proteins, leading to aberrant deacetylation of p53 in inv(16)+ AML cells.14 Notably, that study revealed that HDAC8 mRNA expression is selectively higher in inv(16)+ CD34+ AML cells,14 suggesting the importance of HDAC8 in inv(16)+ LSC transformation and maintenance; however, the mechanism underlying this selectivity remains to be defined. In our study, we found that there are cis elements potentially bound by FOXO1 and FOXO3 at the HDAC8 promoter. Upon FLT3 TKI treatment, both factors increased their binding to the HDAC8 promoter and cooperatively induced HDAC8 transcription. Depletion of FOXO1 and FOXO3 abrogated HDAC8 upregulation and phenocopied HDAC8 inhibition, which could be rescued by HDAC8 overexpression or p53 knockdown.
The finding that FOXO1 and FOXO3 induce HDAC8 expression, which, in turn, deacetylates and inactivates p53, is quite intriguing, because FOXO factors are primarily considered tumor suppressors. However, several recent studies have revealed an important role for FOXO factors in leukemogenesis and LSC maintenance. For example, it is demonstrated that increased FOXO1 induces an oncogenic network in human CD34+ cells and promotes preleukemia transition and that FOXO1 is required by AML1-ETO+ preleukemia cells for the activation of a stem cell molecular program.32 That study also discovered high expression of FOXO1 in inv(16)+ AML samples,32 which may explain the mechanism underlying the selectively higher expression of HDAC8 in inv(16)+ AML LSCs.14 In AML with normal cytogenetics, high FOXO3 expression is associated with a poor prognosis.33 Sykes et al showed that Foxo3 is important for maintaining LSCs in an MLL-AF9 AML mouse model. Foxo3 localized to the nucleus in AML stem cells, and ablation of Foxo3 induced myeloid differentiation and cell apoptosis, leading to reduced leukemia burden and AML LSC frequency.34 In a chronic myeloid leukemia mouse model driven by the constitutively active tyrosine kinase BCR-ABL, Foxo3 shows opposite effects on the survival of LSCs and non-LSCs.35 In non-LSCs, BCR-ABL drives strong Akt activation that forcefully represses Foxo3 functions. In contrast, in LSCs, Akt activity is suppressed despite BCR-ABL expression in vivo, leading to activation and nuclear localization of Foxo3, which sustains the leukemia-initiating capacity. FOXO factors may contribute to the acquisition of dormancy in leukemia cells after exposure to antileukemia agents and allow the LSCs to resist various stresses through a series of mechanisms, such as regulating metabolism,36 inducing a stem cell molecular program,32 reducing oxidative stress,37 and, in our study, inhibiting p53 activity. These studies may help to reconcile the controversy between our findings and the study by Scheijen et al showing that FLT3-ITD promotes Foxo3 phosphorylation and nuclear exclusion and, thus, prevents the induction of Foxo3-mediated apoptosis.38 In that study, FLT3-ITD was ectopically overexpressed in murine Ba/F3 cells, and the interactions between FLT3-ITD and Foxo3 were only investigated with in vitro experiments. A future study using FLT3-ITD+ AML patient samples and a mouse model will be helpful for elucidating the in vivo role of FOXO in FLT3-ITD+ AML LSCs.
HDAC8 upregulation upon FLT3 inhibition was associated with downmodulation of p53 activity in FLT3-ITD+ AML cells. p53 plays a pivotal role in normal and leukemic hematopoiesis. TP53 mutations/loss confer early leukemogenic capacity,39-43 as well as represent a subgroup of AML with the poorest prognosis.3 TP53 mutations occur in only 8% of de novo AML cases2,44 and are mutually exclusive with FLT3-ITD mutations.45 Further studies revealed that, in AML with WT p53, the function of p53 is frequently deregulated through a series of mechanisms, conferring a poor prognosis.46,47 FLT3-ITD mutations may associate with WT p53 dysfunction via SIRT1 overexpression, which yields p53 deacetylation,21,48 AKT and ERK activation, which enhances MDM2-mediated p53 degradation,49 and STAT5 activation, which promotes BCL2 accumulation to oppose p53 activity.50 Theoretically, inhibition of FLT3 would abrogate these mechanisms and enhance the antitumor activity of WT p53 in FLT3-ITD+ AML cells; however, in our study, we found that acetylation of p53 does not show any change upon FLT3 inhibition, and p53 target genes do not show any upregulation either. We propose that the upregulation of HDAC8 counteracts p53 activation, thereby maintaining the survival of FLT3-ITD+ AML under TKI treatment. Indeed, targeting HDAC8 strongly increased the acetylation of p53, which was further enhanced when FLT3 was inhibited simultaneously. Also, targeting HDAC8 together with FLT3 significantly promoted apoptosis of FLT3-ITD+ AML cells and reduced disease burden in leukemia PDX models. A recent report demonstrated that HDAC8 plays a key role in maintaining long-term hematopoietic repopulation via modulating p53 activity under stress, and loss of Hdac8 renders long-term hematopoietic stem cells hypersensitive to genotoxic stress.26 Because AML LSCs share some properties with hematopoietic stem cells, we found that HDAC8 inhibition in the FLT3-ITD+ AML PDX model significantly reduced the frequency of CD34+ human leukemia cells, indicating that targeting HDAC8 can effectively eliminate AML LSCs.
HDAC8 upregulation upon TKI treatment is not restricted to FLT3-ITD+ AML. Using an AML cell line harboring a KITN822K activating mutation and a leukemia mouse model driven by AML1-ETO and KITN822K coexpression, we demonstrated that targeting KIT tyrosine kinase also induced HDAC8 transcription and that combined inhibition of KIT and HDAC8 showed a superior antileukemia effect compared with targeting KIT or HDAC8 alone. Interestingly, a recent study revealed that, in melanoma cells harboring a BRAFV600E mutation, HDAC8 expression was induced under BRAF-MEK inhibitor therapy, and upregulated HDAC8 could protect melanoma cells from TKI treatment.51 Therefore, we speculate that HDAC8 upregulation might be a common mechanism used by cancer cells with activating tyrosine kinase mutations to evade TKI therapy and that a combination of TKIs and HDAC8 inhibitors would be a promising strategy to overcome TKI resistance and achieve sustained remission. More investigations on different kinds of cancers with activating tyrosine mutations, such as EGFR in lung carcinoma, Ras in pancreatic cancer, and PI3K in lymphoma, will be useful for validating our speculation.
In sum, our study identifies a novel FOXO/HDAC8/p53 axis that mediates TKI resistance and promotes leukemia maintenance. Because the tyrosine kinase mutation is 1 of the most frequent genetic aberrations in various cancers, our findings may have an even broader influence in developing new strategies for cancer treatment.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all fellows at the Research Center for Experimental Medicine, Rui Jin Hospital for technical assistance, and all researchers from Shanghai Institute of Hematology for kind advice and the supply of research reagents. The authors are grateful to the patients and their physicians for providing primary specimens for this study.
This work was supported by the National Key R&D Program of China (2017YFA0104502), the Ministry of Science and Technology of China (2016YFE0107200), the National Natural Science Foundation of China (81770187, 81830004, 81770151, 81920108005, 31872842, and 81730007), and the Shanghai Sailing Program (19YF1431500).
Authorship
Contribution: J.L., M.-Y.J., W.-Y.F., and X.-J.C. performed experiments, analyzed the data, and helped to write the manuscript; L.-L.M., Z.-Y.W., and Y.S. performed experiments and helped with animal experiments; R.-F.X. helped with animal experiments; L.-N.W. and L.W. helped to collect primary samples; C.-H.J. and J.-L.J. helped with electrophoretic mobility shift assay experiments; W.-J.Z., Y.-D.S., L.C., W.-H.G., and Y.W. provided advice and reviewed the manuscript; J.-M.L. and D.-L.H. provided material and advice and reviewed the manuscript; and A.-B.L. and J.H. designed the overall concept, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jiong Hu, Department of Hematology, Rui Jin Hospital affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200025, People’s Republic of China; e-mail: hj10709@rjh.com.cn; and Ai-Bin Liang, Department of Hematology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, People’s Republic of China; e-mail: lab7182@tongji.edu.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal