

Key Points
GPRC5D protein is specifically expressed on the surface of plasma cells.
GPRC5DxCD3 bispecific antibody can induce T-cell–mediated killing of GPRC5D+ cells.
Visual Abstract

Professional illustration by Katherine St. John.
Abstract
T-cell–mediated approaches have shown promise in myeloma treatment. However, there are currently a limited number of specific myeloma antigens that can be targeted, and multiple myeloma (MM) remains an incurable disease. G-protein–coupled receptor class 5 member D (GPRC5D) is expressed in MM and smoldering MM patient plasma cells. Here, we demonstrate that GPRC5D protein is present on the surface of MM cells and describe JNJ-64407564, a GPRC5DxCD3 bispecific antibody that recruits CD3+ T cells to GPRC5D+ MM cells and induces killing of GPRC5D+ cells. In vitro, JNJ-64407564 induced specific cytotoxicity of GPRC5D+ cells with concomitant T-cell activation and also killed plasma cells in MM patient samples ex vivo. JNJ-64407564 can recruit T cells and induce tumor regression in GPRC5D+ MM murine models, which coincide with T-cell infiltration at the tumor site. This antibody is also able to induce cytotoxicity of patient primary MM cells from bone marrow, which is the natural site of this disease. GPRC5D is a promising surface antigen for MM immunotherapy, and JNJ-64407564 is currently being evaluated in a phase 1 clinical trial in patients with relapsed or refractory MM (NCT03399799).
Introduction
Multiple myeloma (MM) is a malignant plasma-cell disorder that accounts for ∼10% to 15% of all hematologic cancers.1,2 Treatment options for MM have substantially improved and include the use of proteasome inhibitors, immunomodulatory drugs, monoclonal antibodies, and stem-cell transplantation.3 Despite these therapeutic achievements, MM remains incurable, with significant morbidity and mortality. New therapies are urgently needed to overcome the inevitable resistance to current agents, in particular therapies that address novel targets and/or with new mechanisms of action.
Recent advances in T-cell–mediated therapies suggest that redirecting T cells to specific surface tumor antigens may be an effective means to harness the immune system to induce cancer-cell death and create meaningful and long-lasting clinical responses.4,5 G-protein–coupled receptor class 5 member D (GPRC5D) is a type-C 7-pass transmembrane receptor protein that is predominantly expressed in cells with a plasma-cell phenotype, including the majority of malignant plasma cells from patients with MM.6,7 It is an orphan receptor whose ligand and signaling mechanism are yet to be defined. Levels of GPRC5D messenger RNA (mRNA) expression in patients with MM correlate with plasma-cell burden and genetic aberrations such as Rb-1 deletion and high-risk MM.6 GPRC5D mRNA levels are higher in MM plasma cells than normal cells, and the selective expression of GPRC5D suggests it may represent a potential target for effector-cell–mediated therapy to treat plasma-cell disorders like MM.8,9
To evaluate the therapeutic efficacy of targeting GPRC5D, we developed JNJ-64407564, a humanized bispecific DuoBody antibody that binds to CD3 on T cells and GPRC5D on plasma cells. This antibody was designed to recruit CD3-expressing T cells to GPRC5D-expressing MM cells, resulting in the activation of the T-cell receptor (TCR) signaling pathway. JNJ-64407564 represents a novel therapeutic agent for the treatment of MM and smoldering MM (SMM) and is currently in a phase 1 clinical trial (NCT03399799) in patients with relapsed or refractory MM.
Methods
Cell lines and cell culture
All cell lines used are of human origin and were obtained from either American Type Culture Collection or DSMZ. Cell lines were maintained in a log-phase growth state in RPMI 1640 medium with 10% fetal bovine serum in absence of antibiotics at 37°C in a 5% CO2 incubator and tested to exclude mycoplasma contamination.
Generation of CRISPR knockout (KO) cell lines
A ribonucleoprotein complex was formed by incubation of the control RNA and tracer RNA duplex mixture with Cas9 nuclease and phosphate-buffered saline (PBS) for 20 minutes. H929 cells were electroporated with the ribonucleoprotein complex using single pulses of 165 V for 15 seconds with an ECM830 square wave electroporation system.
GPRC5D mRNA expression in MM patient samples
Bone marrow (BM) aspirate samples from healthy volunteers, patients with premalignant disease (ie, monoclonal gammopathy of undetermined significance and SMM), and patients with malignant disease (ie, MM and plasma-cell leukemia) were enriched for CD138+ cells using immunomagnetic beads (autoMACS; Miltenyi Biotec), and mRNA was analyzed. The Affymetrix GeneChip CEL files were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). Two data sets were evaluated: Agnelli et al10 (GSE16122) and Chng et al11 (GSE6477). For the GeneLogic data set, Affymetrix GeneChip CEL files were obtained from Ocimum Biosolutions. Raw data were processed and normalized independently using the robust multichip averaging method in the Affymetrix Bioconductor R software package.12
Tissue processing and TaqMan analysis
Frozen healthy human tissues were obtained from Cureline. RNA was isolated using RNeasy Mini spin column kit (Qiagen). First-strand synthesis of complementary DNA (cDNA) was generated using the high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). For real-time polymerase chain reaction (PCR), TaqMan Gene Expression Assay (Applied Biosystems) with GPRC5D or glyceraldehyde 3-phosphate dehydrogenase was used in combination with TaqMan PCR Core Reagent Kit (Applied Biosystems). Samples were run using an Applied Biosystems ViiATM 7 qPCR System.
Flow cytometry analysis of GPRC5D expression
Human BM mononuclear cells (MNCs; ProteoGenex) and MM cell lines (1 × 106) were resuspended in LIVE/DEAD staining solution (Life Technologies), incubated for 15 minutes, washed twice, and resuspended in staining mix with 5 µg/mL anti-GPRC5D antibody (571961 clone, R&D Systems) or corresponding isotype (IC0041A) or anti-CD138 (MI15 clone, BioLegend) antibodies. Samples were incubated for 60 minutes, washed twice, resuspended in fluorescence-activated cell sorting (FACS) stain buffer, and analyzed on a BD FACSCanto II cytometer (Becton Dickinson). Data analysis was conducted using FlowJo v 10.1 (Tree Star). For the MM.1S xenograft efficacy study, 3-mm3 tumor samples were processed using GentleMACS and filtered through a 70-µM cell strainer. Cells were processed for staining as described above.
Healthy donor whole blood
Fresh peripheral blood from 3 healthy human donors (Janssen donor program) was incubated with staining antibodies for cell lineage, along with JNJ-64407564, GPRC5DxNull, NullxCD3, or isotype control antibodies. After 30 minutes of incubation, cells were stained with 1:200 LIVE/DEAD near-infrared stain and 1:50 anti-human immunoglobulin G4 (IgG4)-phycoerythrin for 15 minutes, washed, and resuspended in FACS buffer for analysis on an LSR Fortessa (Becton Dickinson). Approximately 100 000 events were collected from each well. Analysis was performed using FlowJo v 10.1+. Cell types were gated using specific markers (B-cell markers: CD3− and CD19+; natural killer (NK)-cell markers: side scatter (SSC)lo, CD3−, CD19−, CD16+, and CD56+; NK T-cell markers: SSClo, CD3+, CD56+, and CD16−; CD4 T-cell markers: CD3+ and CD4+; CD8 T-cell markers: CD3+ and CD8+; monocyte markers: SSChi and CD14+; neutrophil markers: SSChi, CD45+, CD16+, and CD11b+).
Receptor enumeration
Conjugation of anti-GPRC5D antibody (571961 clone) and isotype control (IC0041A) were performed using Lightning-Link R-phycoerythrin (PE) conjugation kit (Innova Biosciences). BD QuantiBRITE PE Beads kit (BD Biosciences) was used to quantify receptor density by measuring the geometric mean fluorescence intensity of GPRC5D staining on cells.
Monoclonal antibody generation
Three BALB/c mice were immunized intradermally with pCMV6-neo plasmid DNA expressing full-length human GPRC5D on days 0, 10, and 20. Mice received final intraperitoneal and IV immunizations on day 59 with rat basophilic leukemia (RBL) cells overexpressing full-length human GPRC5D. On day 63, lymph nodes and spleens were harvested and resulting hybridomas were screened by flow cytometry for binding to both RBL and GPRC5D-overexpressing RBL cell lines. The DuoBody antibody (JNJ-64407564 and JNJ-64024701) was generated (IgG4-S228P, F234A, L235A [PAA] scaffold to minimize Fc binding) by controlled Fab-arm exchange of a GPRC5D antibody (GC5B596 and GC5B673) and an internal CD3 parental antibody following the method developed by Genmab.13 Null arm controls were generated by controlled Fab-arm exchange of GC5B596 with a mouse anti-human respiratory syncytial virus neutralizing antibody (GPRC5DxNull) or the CD3 parental antibody with the mouse anti-human respiratory syncytial virus antibody (NullxCD3).14
T-cell–mediated cytotoxicity and T-cell activation using MM cell lines and healthy donor T cells
Carboxyfluorescein succinimidyl ester (CFSE)-labeled tumor cells were suspended to 2.22 × 105 cells/mL in complete medium containing 2 mg/mL Fc block15 and plated at 2 × 104 cells. Frozen T cells (6 donors; Biological Specialty Corporation; 1 × 105) and antibody (JNJ-64407564, GPRC5DxNull, and NullxCD3) were added and incubated for 48 hours. The supernatant was collected for cytokine analysis, and the cell pellet was stained with LIVE/DEAD stain and phenotyping antibodies (CD3-Pacific blue and CD25-PE). In addition, T-cell proliferation was monitored using and Ki67-Alexa fluor antibody. Samples were analyzed on a FACSCanto II using FlowJo software.
Surface expression and conjugate formation by Amnis imaging
Viable H929, Daudi, and HEK-293 (parental and GPRC5D-overexpressing) cells were stained with internal antibodies against GPRC5D (clone: GC5B483) or the corresponding isotype control (clone: B23B39) in the APC channel and analyzed on Amnis ImageStreamX at 40× magnification; 500 cells were collected per sample. For conjugate formation, healthy control isolated T cells labeled with CellTrace Violet and CellTrace CFSE-labeled target cells (H929 wild-type, B-cell maturation antigen [BCMA] KO, and GPRC5D KO) were mixed at a 5:1 effector/target (E/T) ratio and incubated in the presence or absence of GPRC5DxCD3 DuoBody antibody, fixed with CytoFix (BD Pharmingen), permeabilized with 0.3% Triton X-100 (Sigma), and stained for F-actin using Phalloidin-AF647 (Life Technologies). T-cell/target-cell conjugates were analyzed on Amnis ImageStreamX at 60× magnification; 10 000 total events were collected per sample.
T-cell–mediated cytotoxicity and T-cell activation using H929 target cells and human healthy whole blood
Healthy human whole blood was used as a source of effector T cells, and CFSE-labeled H929 MM cells were spiked-in as target cells at a 5:1 E/T ratio and incubated with antibody for 48 hours. Cell pellets were stained with anti-CD3 and anti-CD25 antibodies, lysed with lysis buffer (BD Biosciences), and stained with LIVE/DEAD stain. After washing, pellets were resuspended in FACS buffer and analyzed by flow cytometry.
T-cell–mediated cytotoxicity and T-cell activation using MM patient CD138+ BM cells and healthy donor T cells
MM patient BM MNCs (ProteoGenex) and T cells were incubated with 100 ng/mL interleukin-6 (IL-6) and JNJ-64407564 or control antibody for 48 hours. Samples were spun and washed; labeled with LIVE/DEAD stain, anti-CD3 and anti-CD25 antibodies, and anti-CD138; and analyzed for CD138+ cells (MI15 antibodies; BioLegend) using a FACSCanto II and FlowJo software.
Cytokine measurement
Production of interferon γ (IFN-γ), IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, and tumor necrosis factor α (TNF-α) was assayed using the Meso Scale Discovery human proinflammatory panel 1 kit as per the instructions (K15049D; Meso Scale Discovery).
Xenograft studies
Female NSG mice (nonobese diabetic severe combined immunodeficiency γ [NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ]; The Jackson Laboratory, Bar Harbor, ME) were used at 6 to 8 weeks of age and weighed ∼20 g. All experiments were carried out in accordance with The Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Janssen R&D, Spring House, PA.
H929 prophylactic model
Mice were injected IV with 1 × 107 human peripheral blood MNCs (PBMCs; donor D204071, lot 14035489, HemaCare) 7 days prior to tumor cell implantation. Seven days after PBMCs were engrafted, each mouse received 5 × 106 H929 cells by subcutaneous (SC) injection in the right flank followed by IV administration of PBS or JNJ-64407564 antibody at 0.1 µg (0.005 mg/kg), 1 µg (0.05 mg/kg), or 10 µg (0.5 mg/kg) per animal on days 0, 3, 5, 7, and 10.
MM.1S established model
Each mouse received 1 × 107 MM.1S human MM cells implanted SC in the right flank on day 0. On day 7, animals received an IV injection of 1 × 107 human PBMCs (donor D204071, lot 15037266, HemaCare). On day 14, mice were randomized to groups with similar starting tumor volume of ∼75 mm3 (range, 72-78 mm3; n = 10/group). On day 15, each mouse received IV administration of PBS or JNJ-64407564 at 0.1 µg (0.005 mg/kg), 1 µg (0.05 mg/kg), 10 µg (0.5 mg/kg), or 50 µg (2.5 mg/kg). Null bispecific antibody controls, GPRC5DxNull and NullxCD3, were each dosed at 10 µg per mouse. Treatments were administered for a total of 7 doses on days 15, 18, 22, 24, 29, 32, and 36.
Blood and tumors were collected. Tumor volume was calculated using the formula tumor volume (mm3) = (a × b2/2), where a represents the length and b the width of the tumor as determined by caliper measurements. Percent tumor growth inhibition (TGI) was defined as the difference between mean tumor volumes of the treated and control group, calculated as % TGI = ((TVc − TVt)/TVc) × 100, where TVc is the mean tumor volume of a given control group and TVt is the mean tumor volume of the treated group. As defined by National Cancer Institute criteria, ≥60% TGI is considered biologically significant.16
In a repeat MM.1S study, JNJ-64407564 at 10 μg/mouse was administered IV, every 3 or 4 days × 3, via the lateral tail vein. Blood and tumors were collected. Half of the resected SC MM.1S tumors was fixed with formalin, and the other half processed for FACS analysis. For immunohistochemistry studies, 4-μm tissue sections were blocked with H2O2 and incubated with primary antibody (CD4 [clone 4B12, Dako] and CD8 [clone SP57, Ventana Medical Systems]) or the IgG isotype control followed by UltraMap anti-rabbit or UltraMap anti-mouse and the ChromoMap Detection Kit (Ventana Medical Systems) or Leica post primary rabbit anti-mouse IgG and anti-rabbit poly horseradish peroxidase IgG (Leica Biosystems).
Data analysis
Analysis was performed using Microsoft Excel (2010) and GraphPad Prism software version 6. Receptor density dot plots were graphed using GraphPad Prism; statistical analysis was performed using unpaired t test with Welch’s correction. Best-fit linear equations, 1-way analysis of variance values, and significance values were calculated in Prism. P values < .05 were considered significant. Histograms were created using FlowJo v 10.1.
Results
GPRC5D protein is expressed on MM cell lines and plasma cells
We analyzed 2 MM patient sample mRNA data sets from the National Center for Biotechnology Information and found GPRC5D to be overexpressed 2 to 4 times in diseased vs normal cells in both data sets (Figure 1A-B). Expression of GPRC5D mRNA in MM patient samples was confirmed using the GeneLogic data set (supplemental Figure 1, available at the Blood Web site). GPRC5D surface protein expression was detected in several MM cancer cell lines (AMO-1, EJM, H929, MM.1R, MOLP-8, and OPM-2) by FACS, but not in cell lines of the myeloid lineage (KG-1 and MOLM-13) or in a B-cell lymphoma cell line (Daudi) (Figure 1C). FACS imaging of the GPRC5D+ H929 cell line (nonpermeabilized) using an Amnis instrument showed a staining pattern consistent with surface expression of GPRC5D (Figure 1D, left). In addition, overexpression of human GPRC5D cDNA in HEK-293 cells showed a similar specific cell-surface staining pattern (Figure 1D, right). GPRC5D protein was also found on CD138+ (plasma-cell marker) BM MNCs from most healthy donors and MM patients by FACS (n = 21 and 23, respectively) using an internally developed anti-GPRC5D monoclonal antibody (mGC5M481; Figure 1E). The mean receptor counts were 1216 ± 155 in normal samples and 1688 ± 413 in MM patient samples and were not statistically different, indicating that GPRC5D is retained on malignant cells, but not overexpressed on average, in contrast to the mRNA, which was elevated compared with normal. To assess the tissue expression profile of GPRC5D expression, we probed 35 healthy human tissues by qPCR and found the highest GPRC5D expression in spleen and lymph nodes, which are known to contain plasma cells, at levels 5× lower than in the H929 MM cell line positive control. Low mRNA expression levels (50-1000× less than H929) were also found in lung, skin, testis, thyroid, and tonsils, and very low but detectable levels were found in salivary gland and brain cerebellum (Figure 1F), consistent with published data.6
![GPRC5D is selectively expressed on the surface of MM cells. (A-B) A bioinformatic analysis of 2 NCI public data sets shows upregulation of GPRC5D mRNA expression on MM patient CD138+ cells as well as SMM and plasma-cell leukemia samples (PCL). mRNA expression was plotted on log2 scale on the y-axis. (C) GPRC5D cell-surface protein expression on MM cells (AMO-1, EJM, H929, MM.1R, MOLP-8, and OPM2), B cells (Daudi), and myeloid cells (KG-1 and MOLM-13) was labeled using an anti-GPRC5D antibody (red line; 571961 clone; R&D Systems) or an isotype control antibody (gray line; IC0041A clone; R&D Systems) for 30 minutes at 4°C and analyzed by flow cytometry. (D) Images of cells with representative distribution of GPRC5D staining on the cell surface are shown. Merged images of GPRC5D staining and bright field are used to evaluate the localization of protein expression on the cell surface. (E) GPRC5D receptor density quantitation of CD138+ BM MNCs from healthy (n = 21) and MM patient samples (n = 23) shows averages of 1600 to 1800 receptors/cell (QuantiBRITE kit; difference is not statistically significant [N.S.]). (F) GPRC5D mRNA levels in various human tissues relative to H929 cells. Samples were analyzed for GPRC5D and glyceraldehyde 3-phosphate dehydrogenase mRNA expression using a TaqMan probe, and relative levels were plotted on the y-axis. Elevated levels of GPRC5D mRNA expression were observed in lymph node and spleen with minimal levels in lung, skin, and testis. The majority of the tissues were negative for GPRC5D. MGUS, monoclonal gammopathy of undetermined significance. GSE, Genomic Spatial Event database.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/15/10.1182_blood.2019003342/1/m_bloodbld2019003342f1.png?Expires=1763473260&Signature=S5MBxtBjAx-MVqXoUa7xVg4VVCM3K3rMlW98QMmxlXS3zQ4x7iFqmIfzlPJuOLdUdFSpk7PgEGEnMJAMXlYokUWq9dF8ly~hQj5kAzF~4n3s76yZVDw~ckpq65912PzYs-Y8hkTGe8rwzikP-rHkQRpsm33etsn93hSQGMQ41u4Jsmuk~-hWFj1qCg7uU9beXApkHHrL7hETi710YkBGtbiCb2QCr2NMoBwtwktisdkydCRF4KigvEQH-vAnJ63lF9R-4LVjAKUK8tQH-ysWny6s0iNhAGX79J0HBzbL~iO1PQzBUr7crh9sAagENdHtENc3V2yk6D9PiIV~1g0snw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
GPRC5D is selectively expressed on the surface of MM cells. (A-B) A bioinformatic analysis of 2 NCI public data sets shows upregulation of GPRC5D mRNA expression on MM patient CD138+ cells as well as SMM and plasma-cell leukemia samples (PCL). mRNA expression was plotted on log2 scale on the y-axis. (C) GPRC5D cell-surface protein expression on MM cells (AMO-1, EJM, H929, MM.1R, MOLP-8, and OPM2), B cells (Daudi), and myeloid cells (KG-1 and MOLM-13) was labeled using an anti-GPRC5D antibody (red line; 571961 clone; R&D Systems) or an isotype control antibody (gray line; IC0041A clone; R&D Systems) for 30 minutes at 4°C and analyzed by flow cytometry. (D) Images of cells with representative distribution of GPRC5D staining on the cell surface are shown. Merged images of GPRC5D staining and bright field are used to evaluate the localization of protein expression on the cell surface. (E) GPRC5D receptor density quantitation of CD138+ BM MNCs from healthy (n = 21) and MM patient samples (n = 23) shows averages of 1600 to 1800 receptors/cell (QuantiBRITE kit; difference is not statistically significant [N.S.]). (F) GPRC5D mRNA levels in various human tissues relative to H929 cells. Samples were analyzed for GPRC5D and glyceraldehyde 3-phosphate dehydrogenase mRNA expression using a TaqMan probe, and relative levels were plotted on the y-axis. Elevated levels of GPRC5D mRNA expression were observed in lymph node and spleen with minimal levels in lung, skin, and testis. The majority of the tissues were negative for GPRC5D. MGUS, monoclonal gammopathy of undetermined significance. GSE, Genomic Spatial Event database.
GPRC5D is selectively expressed on the surface of MM cells. (A-B) A bioinformatic analysis of 2 NCI public data sets shows upregulation of GPRC5D mRNA expression on MM patient CD138+ cells as well as SMM and plasma-cell leukemia samples (PCL). mRNA expression was plotted on log2 scale on the y-axis. (C) GPRC5D cell-surface protein expression on MM cells (AMO-1, EJM, H929, MM.1R, MOLP-8, and OPM2), B cells (Daudi), and myeloid cells (KG-1 and MOLM-13) was labeled using an anti-GPRC5D antibody (red line; 571961 clone; R&D Systems) or an isotype control antibody (gray line; IC0041A clone; R&D Systems) for 30 minutes at 4°C and analyzed by flow cytometry. (D) Images of cells with representative distribution of GPRC5D staining on the cell surface are shown. Merged images of GPRC5D staining and bright field are used to evaluate the localization of protein expression on the cell surface. (E) GPRC5D receptor density quantitation of CD138+ BM MNCs from healthy (n = 21) and MM patient samples (n = 23) shows averages of 1600 to 1800 receptors/cell (QuantiBRITE kit; difference is not statistically significant [N.S.]). (F) GPRC5D mRNA levels in various human tissues relative to H929 cells. Samples were analyzed for GPRC5D and glyceraldehyde 3-phosphate dehydrogenase mRNA expression using a TaqMan probe, and relative levels were plotted on the y-axis. Elevated levels of GPRC5D mRNA expression were observed in lymph node and spleen with minimal levels in lung, skin, and testis. The majority of the tissues were negative for GPRC5D. MGUS, monoclonal gammopathy of undetermined significance. GSE, Genomic Spatial Event database.
A GPRC5DxCD3 antibody can induce T-cell–mediated cytotoxicity of GPRC5D+ cells in vitro
We developed JNJ-64407564, a humanized IgG4 bispecific antibody with IgG4-PAA scaffold that can bind to both GPRC5D on target cells and the ε chain of CD3 on T cells. To assess antibody activity, GPRC5D+ MM cell lines with varying levels of GPRC5D expression (MM.1R [1271 receptors/cell]; OPM-2 [746 receptors/cell], and H929 [191 receptors/cell]) were incubated with purified healthy human T cells at a 5:1 E/T ratio in the presence of JNJ-64407564. JNJ-64407564 promoted efficient T-cell–mediated cytotoxicity of all GPRC5D+ cells (MM.1R 50% effective concentration [EC50] = 0.008 nM; OPM-2 EC50 = 0.15 nM; H929 EC50 = 0.05 nM) but had no effect on the GPRC5D− cell lines NALM-6 and Daudi (Figure 2A). As expected, negative control bispecific antibodies (GPRC5DxNull and NullxCD3) did not exhibit any cytotoxicity. The data points aligned tightly along the generated fit curve with minimal donor-to-donor variability (n = 6 donors). Consistently, JNJ-64407564 induced T-cell activation in the same assays, as evidenced by the increase in CD25 expression on T cells (EC50=0.01 to 0.11 nM), whereas no significant activation was observed when T cells were cultured with JNJ-64407564 and GPRC5D− cells or when control antibodies were incubated with T cells and GPRC5D+ cells (Figure 2B). To further characterize the observed T cell activation, the levels of secreted cytokines in assay supernatants were determined from the in vitro H929 assay described above. JNJ-64407564 treatment led to the secretion of IFN-γ, IL-2, IL-8, IL-10, and TNF-α, consistent with T-cell activation (Figure 2C). IL-1β, IL-4, IL-6, IL-13, and IL-12p70 showed detectable but minimal induction upon T-cell activation (data not shown). In addition, when measured for T-cell proliferation, we observed a dose-dependent response as measured by flow cytometry using Ki67 stain (supplemental Figures 3 and 4).
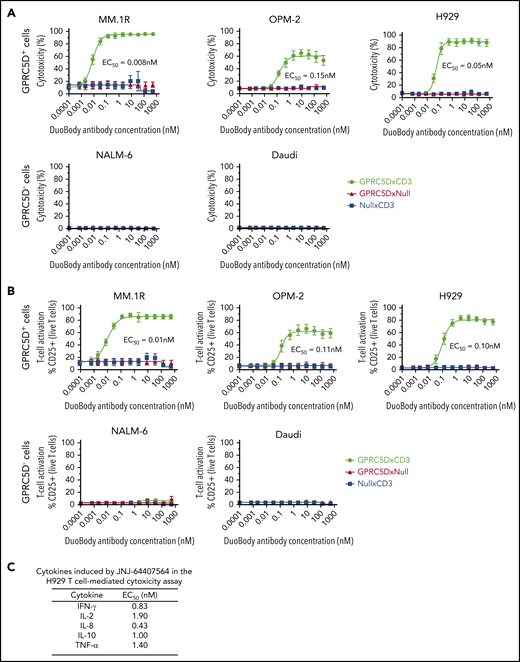
JNJ-64407564 specifically kills GPRC5D+ target cells with concomitant T-cell activation. (A) JNJ-64407564 mediated cytotoxicity of GPRC5D+ cells (MM.1R, OPM-2, and H929) but not GPRC5D− cells (NALM-6 and Daudi), when incubated with healthy purified T cells at a 5:1 E/T ratio. Cytotoxicity was calculated as the percent of remaining CFSC-labeled cells compared with PBS controls after 48-hour incubation with the DuoBody antibodies. JNJ-64407564 (black circles) efficiently killed GPRC5D+ cells, but not GPRC5D− cells, while control antibodies GPRC5DxNull (red triangle) and NullxCD3 (blue square) had no effect. (B) T-cell activation as measured by flow cytometry. T cells were gated using the CD3 surface marker and CD25 activation marker. Percent CD25+ T-cell values were plotted on the y-axis. JNJ-64407564 activated T cells efficiently when incubated with GPRC5D+ cells, but not GPRC5D− cells, while control antibodies GPRC5DxNull and NullxCD3 had no effect. (C) Supernatants from the H929 assays were evaluated for cytokine levels using the Meso Scale Discovery human proinflammatory panel 1 kit (K15049D; Meso Scale Discovery). Average EC50 values from 6 different donor T cells are listed in the table.
JNJ-64407564 specifically kills GPRC5D+ target cells with concomitant T-cell activation. (A) JNJ-64407564 mediated cytotoxicity of GPRC5D+ cells (MM.1R, OPM-2, and H929) but not GPRC5D− cells (NALM-6 and Daudi), when incubated with healthy purified T cells at a 5:1 E/T ratio. Cytotoxicity was calculated as the percent of remaining CFSC-labeled cells compared with PBS controls after 48-hour incubation with the DuoBody antibodies. JNJ-64407564 (black circles) efficiently killed GPRC5D+ cells, but not GPRC5D− cells, while control antibodies GPRC5DxNull (red triangle) and NullxCD3 (blue square) had no effect. (B) T-cell activation as measured by flow cytometry. T cells were gated using the CD3 surface marker and CD25 activation marker. Percent CD25+ T-cell values were plotted on the y-axis. JNJ-64407564 activated T cells efficiently when incubated with GPRC5D+ cells, but not GPRC5D− cells, while control antibodies GPRC5DxNull and NullxCD3 had no effect. (C) Supernatants from the H929 assays were evaluated for cytokine levels using the Meso Scale Discovery human proinflammatory panel 1 kit (K15049D; Meso Scale Discovery). Average EC50 values from 6 different donor T cells are listed in the table.
GPRC5D expression is heterogeneous and independent of BCMA expression
We compared GPRC5D expression in MM patients to that of BCMA, a known MM surface antigen being investigated in various T-cell therapeutics.17 In 51 MM patient BM CD138+ MNC samples, the expression of both proteins was heterogeneous (Figure 3A-B), with most patients displaying a profile comparable to the left panel in Figure 3A. The middle and right panels in Figure 3A show more extreme cases observed in a few patients with dominant expression of one antigen or the other. To confirm the specificity of JNJ-64407564 T-cell–mediated cytotoxicity and determine whether BCMA expression affects GPRC5D expression, we generated isogenic H929 MM cells lacking expression of GPRC5D or BCMA using CRISPR (Figure 3C). While JNJ-64407564 was able to kill wild-type H929 cells, the T-cell–mediated cytotoxicity was abrogated in the presence of GPRC5D− H929 cells, demonstrating the specificity of the antibody (Figure 3D; supplemental Figure 2). KO of BCMA did not affect GPRC5D expression, and BCMA− H929 cells could be killed by JNJ-64407564 (Figure 3C-D; supplemental Figure 2). KO of GPRC5D did not affect the expression of BCMA, suggesting that these 2 proteins are regulated independently, which is consistent with the heterogenous expression seen in Figure 3B. A similar experiment using a tool BCMAxCD3 antibody showed that it could kill MM cells expressing BCMA, whether GPRC5D was present or not (Figure 3D).
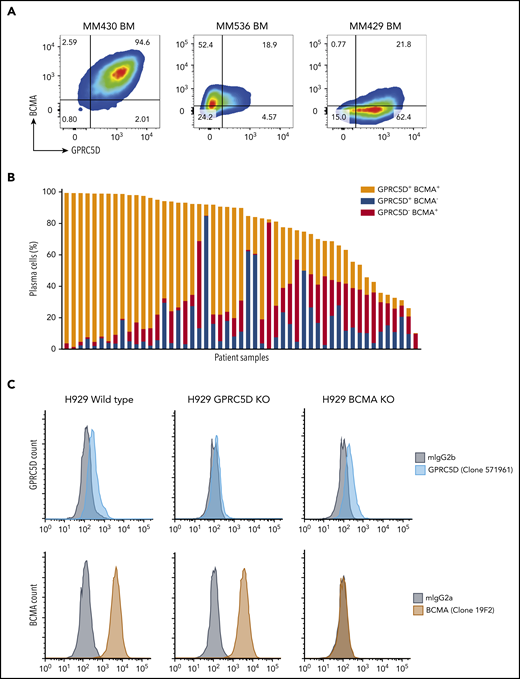
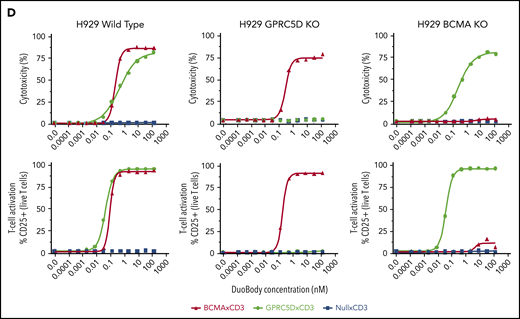
Expression of GPRC5D and BCMA on MM cells. (A) MM patient BM MNCs showing various profiles of GPRC5D and BCMA expression. Cells were gated using the CD138 plasma-cell marker (MI15; BioLegend) and antibodies to GPRC5D and BCMA to measure their surface expression (GPRC5D: 571961, R&D Systems; BCMA: 19F2, BioLegend). The left panel is representative of most patient samples surveyed, while the middle and right panels depict skewed expression for BCMA or GPRC5D, which was seen in a minority of patients (see panel B). (B) Expression of GPRC5D and BCMA surface protein in 51 BM MNC samples from MM patients on CD138+ cells as measured by flow cytometry. Each column represents an individual patient sample. Data are expressed as percent positivity of all CD138+ cells. (C) Generation of GPRC5D (top row) and BCMA (bottom row) KO H929 cells by CRISPR. Loss of signal can be seen in the KO cells compared with the isotype control (gray). (D) JNJ-64407564 (black circle) efficiently depleted H929 wild-type or BCMA KO cells, but not GPRC5D KO cells. Percent cytotoxicity and T-cell activation were measured as in Figure 2. A positive control BCMAxCD3 antibody (red triangle) killed wild-type H929 and GPRC5D KO cells but had no effect on BCMA KO cells. The negative control antibody NullxCD3 (blue square) had no effect on cytotoxicity or T-cell activation in all cell lines.
Expression of GPRC5D and BCMA on MM cells. (A) MM patient BM MNCs showing various profiles of GPRC5D and BCMA expression. Cells were gated using the CD138 plasma-cell marker (MI15; BioLegend) and antibodies to GPRC5D and BCMA to measure their surface expression (GPRC5D: 571961, R&D Systems; BCMA: 19F2, BioLegend). The left panel is representative of most patient samples surveyed, while the middle and right panels depict skewed expression for BCMA or GPRC5D, which was seen in a minority of patients (see panel B). (B) Expression of GPRC5D and BCMA surface protein in 51 BM MNC samples from MM patients on CD138+ cells as measured by flow cytometry. Each column represents an individual patient sample. Data are expressed as percent positivity of all CD138+ cells. (C) Generation of GPRC5D (top row) and BCMA (bottom row) KO H929 cells by CRISPR. Loss of signal can be seen in the KO cells compared with the isotype control (gray). (D) JNJ-64407564 (black circle) efficiently depleted H929 wild-type or BCMA KO cells, but not GPRC5D KO cells. Percent cytotoxicity and T-cell activation were measured as in Figure 2. A positive control BCMAxCD3 antibody (red triangle) killed wild-type H929 and GPRC5D KO cells but had no effect on BCMA KO cells. The negative control antibody NullxCD3 (blue square) had no effect on cytotoxicity or T-cell activation in all cell lines.
GPRC5DxCD3 antibody can induce T-cell–mediated cytotoxicity of GPRC5D+ cells in healthy human whole blood
To characterize the activity of JNJ-64407564 in a more physiologic environment, the antibody was tested in 2 ways. First, JNJ-64407564 was added to healthy human whole blood in the presence of GPRC5D+ H929 cells, since whole blood normally contains very few plasma cells. JNJ-64407564 was active in killing GPRC5D+ cells in all samples (6/6 donors) (Figure 4A-B), and this activity correlated with T-cell activation (cytotoxicity EC50 = 0.20-0.61 nM; T-cell activation EC50 = 0.12-0.40 nM). The activity of JNJ-64407564 was significantly decreased in whole blood compared with the in vitro purified system described above (∼20-200× change at 48 hours), which could be due to the complexity of the whole blood matrix or slower killing kinetics. The addition of JNJ-64407564 alone to whole blood did not induce T-cell activation due to lack or low levels of circulating plasma cells (Figure 4C). Binding studies by FACS also showed that JNJ-64407564 only bound to T and NK T cells in whole blood as expected due to the presence of CD3ε on these cells (Figure 4D) and support the observed lack of cytotoxicity in the absence of H929 cells (data not shown).
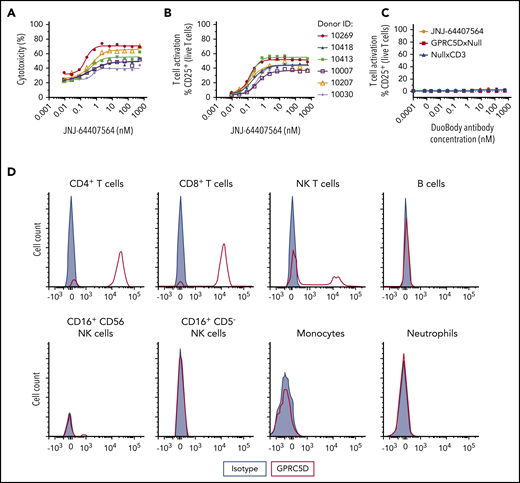
JNJ-64407564 can deplete MM cells when incubated with healthy human whole blood. (A-B) Human MM cells (H929) were incubated for 48 hours with whole blood from 6 different healthy donors at a 5:1 E/T ratio in the presence of various concentrations of JNJ-64407564. Percent cytotoxicity and T-cell activation were calculated as in Figure 2. (C) Healthy whole blood was incubated with JNJ-64407564 (black circle), GPRC5DxNull (white square), and NullxCD3 (black triangle) for 48 hours without exogenous target cells. (D) Binding of JNJ-64407564 to different cell populations in healthy human whole blood. The antibody was incubated with whole blood at 4°C for 30 minutes, and the binding profile was measured by flow cytometry (JNJ-64407564, red trace; IgG4-PAA isotype control, gray filled). Binding was captured using a PE-labeled secondary antibody (HP6025; SouthernBiotech).
JNJ-64407564 can deplete MM cells when incubated with healthy human whole blood. (A-B) Human MM cells (H929) were incubated for 48 hours with whole blood from 6 different healthy donors at a 5:1 E/T ratio in the presence of various concentrations of JNJ-64407564. Percent cytotoxicity and T-cell activation were calculated as in Figure 2. (C) Healthy whole blood was incubated with JNJ-64407564 (black circle), GPRC5DxNull (white square), and NullxCD3 (black triangle) for 48 hours without exogenous target cells. (D) Binding of JNJ-64407564 to different cell populations in healthy human whole blood. The antibody was incubated with whole blood at 4°C for 30 minutes, and the binding profile was measured by flow cytometry (JNJ-64407564, red trace; IgG4-PAA isotype control, gray filled). Binding was captured using a PE-labeled secondary antibody (HP6025; SouthernBiotech).
GPRC5DxCD3 antibody can induce T-cell–mediated cytotoxicity of plasma cells (CD138+ and GPRC5D+) in MM patient samples
In the second approach, JNJ-64407564 was added to MNCs from freshly thawed BM MNCs from MM patients supplemented with exogenous T cells from healthy donors. JNJ-64407564 could bind and deplete primary plasma cells from 4 MM patients in a concentration-dependent manner, as evidenced by the loss of CD138+ cells (top and middle rows, respectively) (Figure 5). The GPRC5D receptor density in these patient samples ranged between 252 and 16 471 receptors per cell as measured by QuantiBRITE beads (data not shown). T-cell activation was observed concomitantly with the cytotoxicity (mean EC50 values for killing = 0.13 ± 0.16 nM; mean EC50 values for T-cell activation = 0.06 ± 0.05 nM). Low or no cytotoxicity or T-cell activation was seen when using control null antibodies. Thus, JNJ-64407564 could efficiently deplete primary CD138+ cells in BM MM samples with various levels of GPRC5D surface expression.
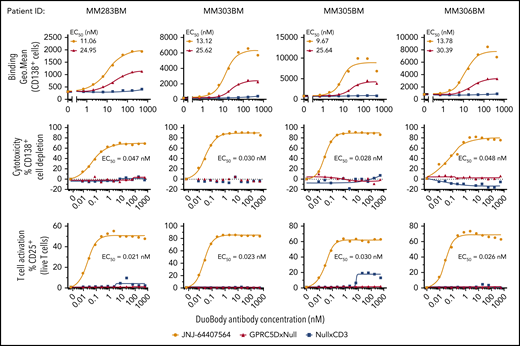
JNJ-64407564 can bind and deplete primary MM patient BM CD138+ cells. Frozen BM MNCs were incubated with various concentrations of JNJ-64407564 (0-532 nM) with or without exogenous healthy T cells to measure target binding and killing. Dose-dependent binding of JNJ-64407564 (black circles) and GPRC5DxNull (blue square) to target cells (top). NullxCD3 (red triangle) failed to show any binding. Dose-dependent plasma-cell depletion (middle). BM MNCs were incubated for 48 hours with exogenous healthy T cells at a 1:1 E/T ratio in the presence of JNJ-64407564 and depletion measured as remaining CD138+ and BCMA+ cells. JNJ-64407564-mediated T-cell activation was measured by flow cytometry by gating T cells using CD3 surface marker and CD25 activation marker (bottom). Percent CD25+ T-cell values were plotted on the y-axis. JNJ-64407564 (circles) was able to activate T cells efficiently when incubated with GPRC5D+ cells, but not GPRC5D− cells, while control antibodies GPRC5DxNull (triangle) and NullxCD3 (square) had no effect.
JNJ-64407564 can bind and deplete primary MM patient BM CD138+ cells. Frozen BM MNCs were incubated with various concentrations of JNJ-64407564 (0-532 nM) with or without exogenous healthy T cells to measure target binding and killing. Dose-dependent binding of JNJ-64407564 (black circles) and GPRC5DxNull (blue square) to target cells (top). NullxCD3 (red triangle) failed to show any binding. Dose-dependent plasma-cell depletion (middle). BM MNCs were incubated for 48 hours with exogenous healthy T cells at a 1:1 E/T ratio in the presence of JNJ-64407564 and depletion measured as remaining CD138+ and BCMA+ cells. JNJ-64407564-mediated T-cell activation was measured by flow cytometry by gating T cells using CD3 surface marker and CD25 activation marker (bottom). Percent CD25+ T-cell values were plotted on the y-axis. JNJ-64407564 (circles) was able to activate T cells efficiently when incubated with GPRC5D+ cells, but not GPRC5D− cells, while control antibodies GPRC5DxNull (triangle) and NullxCD3 (square) had no effect.
JNJ-64407564 prevents tumor growth and regresses established tumors in human MM xenograft models
To assess the activity of JNJ-64407564 in vivo, we used 2 tumor models of MM. In the prophylactic H929 model, JNJ-64407564 completely prevented tumor formation at both 1 µg and 10 µg/mouse doses (TGI = 100%, P ≤ .05; Figure 6A). In the MM.1S model, statistically significant antitumor activity was observed at 10 and 50 µg/mouse doses, with 10 out of 10 mice showing complete responses (tumor regressions) in each group (Figure 6B). The 1 µg/mouse dose significantly inhibited tumor growth by 65% (P ≤ .05) as compared with PBS-treated control animals. The negative control antibodies, NullxCD3 and GPRC5DxNull, did not suppress tumor growth (Figure 6B).
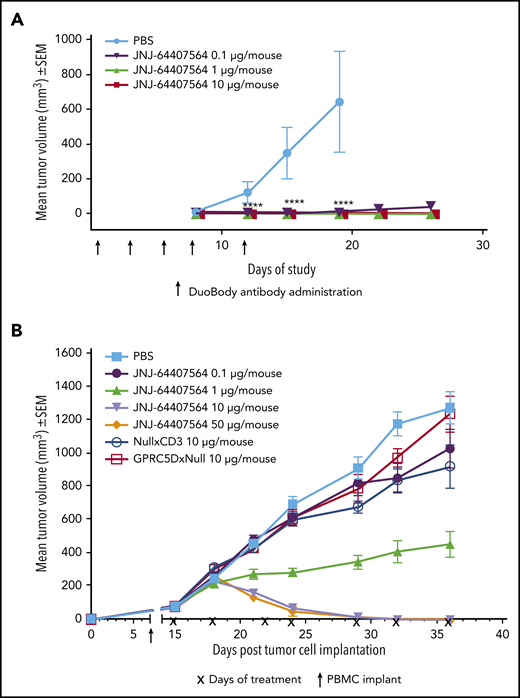
JNJ-64407564 can inhibit and regress tumors in murine models of MM. (A) JNJ-64407564 inhibits H929 tumor growth in a prophylactic xenograft model. NSG mice were injected IV with 1 × 107 human PBMCs, and after 7 days, each mouse received SC injection of 5 × 106 H929 cells in the right flank followed by IV administration of PBS (black circles) or JNJ-64407564 antibody at 0.1 µg (0.005 mg/kg, green downward triangle), 1 µg (0.05 mg/kg, red upward triangle), or 10 µg (0.5 mg/kg, blue square) per animal on days 0, 3, 5, 7, and 10. By day 19, the mean tumor volume of the PBS-treated control group (n = 6) had exceeded 600 mm3, and the mice were terminated. (B) JNJ-64407564 causes tumor regression in MM.1S MM xenograft model. Each mouse received 1 × 107 MM.1S cells in PBS in a total volume of 0.2 mL. Cells were implanted SC in the right flank using a 1-cm3 syringe and a 26-gauge needle. The day of tumor cell implantation was designated as day 0. On day 7 after tumor cell implant, animals were randomized with a tumor volume of ∼75 mm3 and received IV injection of 1 × 107 human PBMCs. Treatments were initiated on day 15, with each mouse receiving IV administration of PBS (black square) or JNJ-64407564 at 0.1 µg (0.005 mg/kg, blue circle), 1 µg (0.05 mg/kg, green upward triangle), 10 µg (0.5 mg/kg, red downward triangle), or 50 µg (2.5 mg/kg, orange diamond). Null bispecific antibody controls, GPRC5DxNull (dark green open square) and NullxCD3 (purple open circle), were each dosed at 10 µg per mouse. Treatments were administered for a total of 7 doses on days 15, 18, 22, 24, 29, 32, and 36 (indicated as “x” on the x-axis). Tumor measurements were taken up to day 36, when 80% to 100% of mice remained on study for each group. SEM, standard error of the mean.
JNJ-64407564 can inhibit and regress tumors in murine models of MM. (A) JNJ-64407564 inhibits H929 tumor growth in a prophylactic xenograft model. NSG mice were injected IV with 1 × 107 human PBMCs, and after 7 days, each mouse received SC injection of 5 × 106 H929 cells in the right flank followed by IV administration of PBS (black circles) or JNJ-64407564 antibody at 0.1 µg (0.005 mg/kg, green downward triangle), 1 µg (0.05 mg/kg, red upward triangle), or 10 µg (0.5 mg/kg, blue square) per animal on days 0, 3, 5, 7, and 10. By day 19, the mean tumor volume of the PBS-treated control group (n = 6) had exceeded 600 mm3, and the mice were terminated. (B) JNJ-64407564 causes tumor regression in MM.1S MM xenograft model. Each mouse received 1 × 107 MM.1S cells in PBS in a total volume of 0.2 mL. Cells were implanted SC in the right flank using a 1-cm3 syringe and a 26-gauge needle. The day of tumor cell implantation was designated as day 0. On day 7 after tumor cell implant, animals were randomized with a tumor volume of ∼75 mm3 and received IV injection of 1 × 107 human PBMCs. Treatments were initiated on day 15, with each mouse receiving IV administration of PBS (black square) or JNJ-64407564 at 0.1 µg (0.005 mg/kg, blue circle), 1 µg (0.05 mg/kg, green upward triangle), 10 µg (0.5 mg/kg, red downward triangle), or 50 µg (2.5 mg/kg, orange diamond). Null bispecific antibody controls, GPRC5DxNull (dark green open square) and NullxCD3 (purple open circle), were each dosed at 10 µg per mouse. Treatments were administered for a total of 7 doses on days 15, 18, 22, 24, 29, 32, and 36 (indicated as “x” on the x-axis). Tumor measurements were taken up to day 36, when 80% to 100% of mice remained on study for each group. SEM, standard error of the mean.
JNJ-64407564 promoted a decrease in BCMA+/CD38+ tumor cells 24 hours after the first dose (Figure 7A). This BCMA antibody was used for 2 reasons: (1) as a second plasma-cell marker, and (2) since the sample had been pretreated with GPRC5DxCD3 (in vivo), we wanted to avoid using a GPRC5D antibody in the ex vivo testing.
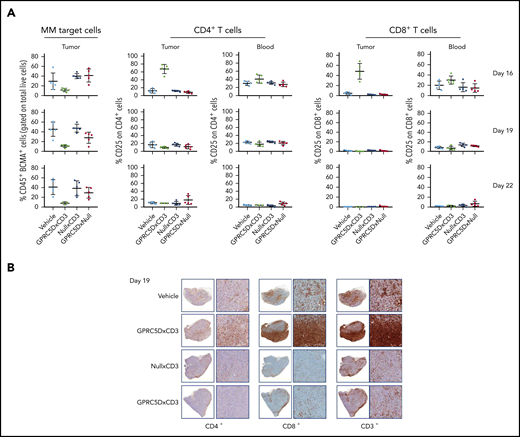
GPRC5D+ cells are depleted from MM.1S tumors. (A-B) A repeat MM.1S study was conducted testing 1 dose (10 μg) of JNJ-64407564 and control bispecific antibodies. (A) Tumor and blood samples were analyzed on the day after dosing on days 16 and 19 for tumor-cell number by staining with anti-BCMA antibody (percent remaining human CD45+ and BCMA+ cells on the y-axis; first column) and T-cell activation (percent CD25+ and CD4+ or CD8+ T cells on the y-axis; remaining columns). The BCMA antibody was used as a second plasma-cell marker to avoid using a GPRC5D antibody in the ex vivo testing. (B) T-cell (CD3+, CD4+, CD8+; SP57, Ventana) infiltration within the tumors. For each section, the left panel is low magnification (1.5×) and the right panel is a higher magnification (10×).
GPRC5D+ cells are depleted from MM.1S tumors. (A-B) A repeat MM.1S study was conducted testing 1 dose (10 μg) of JNJ-64407564 and control bispecific antibodies. (A) Tumor and blood samples were analyzed on the day after dosing on days 16 and 19 for tumor-cell number by staining with anti-BCMA antibody (percent remaining human CD45+ and BCMA+ cells on the y-axis; first column) and T-cell activation (percent CD25+ and CD4+ or CD8+ T cells on the y-axis; remaining columns). The BCMA antibody was used as a second plasma-cell marker to avoid using a GPRC5D antibody in the ex vivo testing. (B) T-cell (CD3+, CD4+, CD8+; SP57, Ventana) infiltration within the tumors. For each section, the left panel is low magnification (1.5×) and the right panel is a higher magnification (10×).
A concomitant increase in activated T cells (CD25+, CD4+, and CD8+ T cells) was also observed in tumor samples and not in the blood after the first dose (day 16; Figure 7B). PBS and null controls did not show reduction of MM.1S tumor cells or activation of T cells (CD25+ on CD4+ or CD8+; Figure 7A-B) in tumor or circulating blood. BCMA+ cells remained depleted after the second dose (day 19; Figure 7A, bottom row), while T-cell activation was not detected, suggesting that target cells were no longer present. Additionally, there was a significant increase in T-cell infiltration in tumors from animals treated with JNJ-64407564, as seen by immunohistochemistry staining of tumor fragments. As shown in Figure 7B, a qualitative increase in CD3+, CD8+, and CD4+ cells in the tumors was observed in the JNJ-64407564–treated samples as compared with the PBS and control antibody-treated samples at day 19.
Toxicology
Pivotal toxicology study conducted with a surrogate molecule JNJ-64024701 (for hazard identification) did not identify any robust pharmacodynamic effect or toxicological response in cynomolgus monkeys (supplemental Table 1).
Discussion
Bispecific antibodies that can bind simultaneously to the TCR coreceptor CD3 on T cells and to specific antigens on tumor cells are attractive therapeutics, as they can promote T-cell–mediated tumor lysis, circumventing the major histocompatibility complex restriction of the TCR. This promising approach has been validated in the clinic with the regulatory approval of blinatumomab for acute lymphoblastic leukemia.18 The potential success of CD3 bispecific approaches relies on the presence of antigens specifically found on the surface of tumor cells. Our results show that GPRC5D protein is expressed on the surface of MM cell lines (FACS and imaging) and patient primary samples by FACS (nonpermeabilized cells), clearly establishing GPRC5D as a surface antigen.
GPRC5D protein expression on tumor cells from MM patients is heterogeneous but present in all 51 patients surveyed. Early analyses suggest that GPRC5D may be expressed in myeloma precursor states monoclonal gammopathy of undetermined significance and SMM. The limited expression in normal tissues including heme cells (except for plasma cells) makes it an attractive target for T-cell therapy. Additionally, since GPRC5D is a GPCR, the exposed epitopes are likely proximal to the plasma membrane, facilitating tighter immunological synapses between T cells and target cells which could drive greater cytotoxicity.19 Furthermore, because GPRC5D is a 7-pass transmembrane protein, it is unlikely to be shed in the serum, a phenomenon seen with other surface antigens such as BCMA20 that could lead to a sink effect and reduced efficacy.21 JNJ-64407564 induced highly specific in vitro T-cell–mediated cytotoxicity against MM cell lines expressing different levels of GPRC5D receptors per cell (191 to 1271 receptors per cell) and also induced T-cell–mediated killing of CD138+ cells in primary BM samples from both newly diagnosed or relapsed/refractory MM patients. JNJ-64407564 treatment resulted in profound tumor regression (10/10) in the established MM.1S xenograft model, with activity correlated with the infiltration of T cells in the tumor as predicted by the mode of action. The pivotal toxicology cynomolgus monkey study conducted using a surrogate molecule (JNJ-64024701) showed no adverse findings.
BCMA is a well-known plasma-cell antigen that is being targeted using several therapeutic modalities such as chimeric antigen receptor T cells, antibody-toxin conjugates, and bispecific antibodies.21-23 While these therapeutics have shown good clinical responses,24 relapses occur in part due to the loss of the BCMA antigen,25 highlighting the need for new antigens in this indication. Our results show that GPRC5D expression is unaffected by the loss of BCMA in H929 MM cells. Indeed, these cells can be killed effectively by JNJ-64407564, supporting the use of this therapeutic antibody against eventual anti-BCMA therapy-resistant clones or in patients with low or absent BCMA expression. Furthermore, our data show that MM patient plasma cells exhibit differential expression of both BCMA and GPRC5D. These data are consistent with a recent report suggesting that BCMA and GPRC5D may be independently regulated in MM plasma cells9 and argue that GPRC5D may represent an attractive target for MM. The lack of GPRC5D expression in memory B cells and progenitor cells further suggests that the humoral immune function should not be significantly affected by treatment with JNJ-64407564.The therapeutic potential of JNJ-64407564 is currently being evaluated in a phase 1 clinical trial in patients with relapsed/refractory MM (NCT03399799).
The data-sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. Requests for access to the study data can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Emily Chen and Anna Hughes for technical help and Tracy Cao for help with the submission; Rachel Goldsmith, Chidozie Amuzie, Brandon Miller, and Meredith Rocca for technical, operational, and scientific support of the nonclinical safety program; and Joanne Ma and Suzette Girgis for pharmacokinetic studies. The authors also thank Brendan Weiss, Blake J. Bartlett, Mike Kelley, Jacintha Shenton, and Jennifer Smit for critical reading of the manuscript.
Authorship
Contribution: K. Pillarisetti, R.A., and F.G. conceptualized the research and formed the hypothesis of this paper; K. Pillarisetti and S.E. designed and performed experiments and analyzed data; S.E., M.T., and N.M. designed and performed antibody generation work; K. Pillarisetti, M.M., Y.L., A.B., D.R., J.J., and M.H. performed the in vitro research and collected data; E.W., K. Pillarisetti, and M.M. generated the CRISPR cell lines; L.L., D.C., and K. Packman designed, performed, and analyzed animal studies; S.S. designed and analyzed the toxicology study; K. Pillarisetti and F.G. wrote the first draft of the manuscript; and all authors critically evaluated and edited the manuscript.
Conflict-of-interest disclosure: All authors are employees of Janssen Pharmaceuticals and have ownership interest in Johnson & Johnson.
Correspondence: François Gaudet, Janssen Research & Development, 1400 McKean Rd, Spring House, PA 19477; e-mail: fgaudet@its.jnj.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal