

In this issue of Blood, report on how novel mutations in the transmembrane (TM) domain of the thrombopoietin (TPO) receptor cause familial essential thrombocythemia and intriguingly link their potential mechanisms of action with that of receptor agonists.1
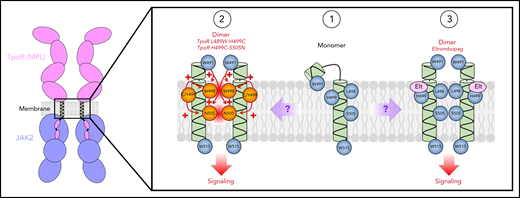
Comparative mechanisms of action between TM mutations in familial ET and Elt proposed in this issue of Blood by Levy and colleagues. In the monomeric state, (1) a possible break in the secondary helical structure of the TM domain may result in W491 associating on the extracellular side of the plasma membrane. TM mutations seen in familial ET (2) may lead to the rotation on W491 inward, stabilizing interactions between other TM residues leading to activation. The authors also propose that Elt may use a similar mechanism (3) via interactions with H499 and W491, breaking the TpoR-membrane interactions and permitting receptor activation.
Comparative mechanisms of action between TM mutations in familial ET and Elt proposed in this issue of Blood by Levy and colleagues. In the monomeric state, (1) a possible break in the secondary helical structure of the TM domain may result in W491 associating on the extracellular side of the plasma membrane. TM mutations seen in familial ET (2) may lead to the rotation on W491 inward, stabilizing interactions between other TM residues leading to activation. The authors also propose that Elt may use a similar mechanism (3) via interactions with H499 and W491, breaking the TpoR-membrane interactions and permitting receptor activation.
The hematopoietic cytokine TPO is the primary regulator of megakaryopoiesis, driving both megakaryocyte differentiation and progenitor expansion, as well as having essential roles in hematopoietic stem cell survival and proliferation.2 The TPO receptor, TpoR (also known widely as MPL), is a member of the type I cytokine receptor family, sharing sequence and functional homologies with the receptors for erythropoietin, prolactin, and growth hormone.3 In addition to its key roles in physiological hematopoiesis, TpoR has emerged as the common mechanistic link between the prevalent driver mutations in the Philadelphia chromosome negative myeloproliferative neoplasms (MPNs). Mutated JAK2 and CALR interact with TpoR and are both reliant on the expression of physiological levels of the receptor to cause disease,4 whereas a small number of MPN patients harbor mutations in TpoR itself, primarily in the TM and intracellular juxtamembrane (JM) regions (S505N and W515K, respectively), directly leading to receptor hyperactivity.5,6 Gaining a complete understanding of how TpoR mutations alter receptor activity not only is important clinically but also provides a fascinating insight into cytokine receptor function, especially the role of the plasma membrane and membrane-associated regions of the receptor.
In this issue of Blood, Levy et al identify 2 familial essential thrombocythemia (ET) patients with novel double mutations of TpoR, L498W-H499C and H499Y-S505N, which highlight how subtle interactions between amino acids in the TM can result in potent changes in receptor activity (see figure). To understand the functional activity of these mutations, the authors generated cell lines expressing the TpoR mutations alone and in combination. The L498W mutation increases cytokine-independent STAT5 activity and cellular proliferation, both of which are potentiated by expressing L498W-H499C in combination, suggesting that H499C acts additively with the L498W mutation. As TpoR needs to form dimers to allow the cross-phosphorylation and activation of associated JAK2 molecules, the authors have applied a luciferase complementation assay that enables them to estimate the proportion of receptors that are in a dimeric state in the presence or absence of the newly identified mutations. Interestingly, both L498W and H499C increase the proportion of dimeric TpoR both alone and in combination; however, given the lack of factor-independent signaling and growth in cells expressing H499C, it would appear that in this case the dimeric complex fails to initiate signal transduction. Furthermore, both L498W and H499C are able to initiate signaling in the absence of the extracellular domain (truncated at position 489), suggesting that these mutations work by altering the configuration of the TM and cytosolic domains to allow interactions between the associated JAK2 molecules.
TpoR H499Y, which was found in combination with S505N in the second patient in this study, was also able to activate STAT5 and greatly increased S505N activity when expressed together. However, H499Y was unable to further enhance L498W activity while mutating either L498 or H499 enhanced signaling by the more common TpoR W515K mutation, highlighting the diversity in the mechanisms of action between mutations in the TpoR TM and intracellular JM regions.
Throughout the study, the authors draw potential parallels between the TM mutations and the mechanism of eltrombopag (Elt), a small molecule TpoR agonist used for the treatment of idiopathic thrombocytopenia purpura.7 Although TpoR-H499 is essential for Elt activity,8 it has been suggested previously that other amino acids upstream of H499 are equally important. A lack of Elt activity on the truncated TpoR supports this theory and leads the authors to investigate potentially important amino acids in the extracellular JM region of the receptor. Taking a structural modeling approach, W491 was identified as a key upstream residue. Being located on the same helical face as L498, S505, and W515 in the active dimer, the authors hypothesize that altering the position of W491 by mutating the tryptophan to an alanine or lysine may change the TM structure and reduce the activity of the L498W, S505N, and W515K mutations and potentially Elt. This was confirmed by generating a series of cell lines expressing TpoR with a combination of point mutations, elegantly demonstrating that introducing TpoR W491A completely abrogates the response to Elt and the activity of the TM and W515 mutations. Together, this raises the exciting potential of shared mechanisms of action between Elt and TpoR activating mutations. Moreover, as W491 is most likely more accessible in the JM region compared with the TM mutations, and it also raises the possibility that this residue could be targeted clinically to reduce the activity of TpoR mutations.
Despite these interesting insights into TpoR activity, key questions persist. A complete structure of TpoR remains elusive and continues to hinder our understanding of how the receptor is activated, both physiologically by TPO and in the case of MPN driver mutations. This is further complicated by the fact that JAK2 will undoubtedly play a critical role in receptor stabilization, dimerization, localization, and interactions with the plasma membrane, so a structure of the receptor alone will not provide answers to all the key questions. Moreover, receptor density at the plasma membrane will alter the ability of the activating mutations to drive receptor dimerization, highlighting one of the issues faced by all of us using TpoR-overexpressing cell lines to model receptor activity. Clearly, this fascinating receptor still has many secrets left for us to discover.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal