

In this issue of Blood, report that interferon regulatory factor 4 (IRF4) and nuclear factor κ light chain enhancer of activated B cells (NF-κB) drive maintenance of adult T-cell leukemia/lymphoma (ATL) by coordinately stimulating a transcriptional regulatory network normally intended for promoting T-cell immune functions.1
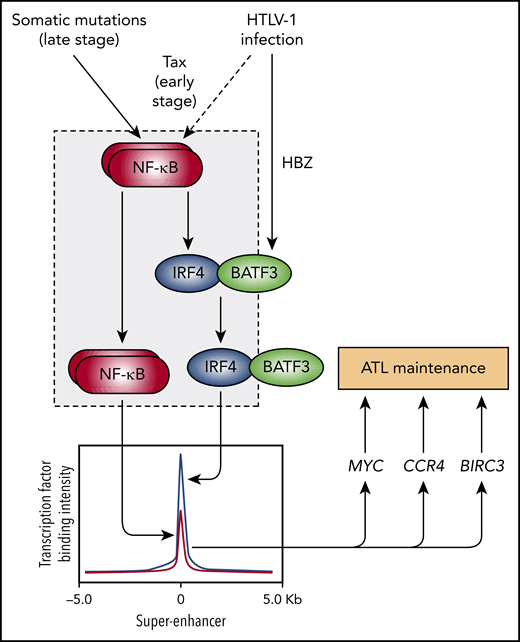
Diagram depicting a network of transcriptional interactions driving ATL maintenance that is initiated by HTLV-1 infection and then propagated by somatic mutations. The gray box indicates the network motif identified by Wong et al called a “coherent feed-forward loop” that is used by normal T cells to rapidly expand to fight foreign invaders but is hijacked and constitutively activated in ATL cells. This loop consists of the top-tier transcription factor NF-kB inducing the second-tier transcription factor IRF4 followed by cooperative binding and activation by the 2 factors of super-enhancers associated with major oncogenes (MYC, CCR4, and BIRC3).
Diagram depicting a network of transcriptional interactions driving ATL maintenance that is initiated by HTLV-1 infection and then propagated by somatic mutations. The gray box indicates the network motif identified by Wong et al called a “coherent feed-forward loop” that is used by normal T cells to rapidly expand to fight foreign invaders but is hijacked and constitutively activated in ATL cells. This loop consists of the top-tier transcription factor NF-kB inducing the second-tier transcription factor IRF4 followed by cooperative binding and activation by the 2 factors of super-enhancers associated with major oncogenes (MYC, CCR4, and BIRC3).
Despite carrying few genes, viruses can reshape the genetic landscape of normal cells and can start the process of transforming those cells into cancer cells through diverse mechanisms. A classic example is human T-cell lymphotropic virus type 1 (HTLV-1), which integrates into the genome of mature T cells and pushes them into becoming ATL cells.2 The few proteins encoded by the limited HTLV-1 genome must cleverly hijack the normal cellular machinery of T cells so that HTLV-1 can thrive and reproduce. In T cells, the NF-κB pathway is primed to activate a transcriptional regulatory network of genes to drive cell proliferation in response to pathogens and other dangers. Uninfected T cells switch the NF-κB pathway on and off as needed. In contrast, HTLV-1–infected T cells express an oncoprotein called Tax that flips the switch to the on position.2 If the infected T cells acquire mutations in the NF-κB pathway, the switch is flipped permanently to the on position, transforming precancerous cells into fully malignant ATL cells.3 Fortunately, this happens in only about 5% of patients after more than 50 years of viral latency. But when ATL happens, the consequences can be devastating. Median survival is less than 1 year.
To improve outcomes, investigators have sought to better understand the ATL cancer drivers in the hopes of finding new vulnerabilities that might someday be targeted by drugs. Nakagawa et al4 showed that a virally encoded oncoprotein called HTLV-1 basic leucine zipper factor (HBZ) induces the expression of the basic leucine zipper ATF-like transcription factor 3 (BATF3). BATF3 has a partner called IRF4 that is highly expressed and can be somatically mutated in ATL cells, driving T-cell proliferation.3,5 Together, BATF and IRF factors form a transcription factor complex that induces genes important for various T-cell immune functions,6 including MYC, one of the most essential ATL oncogenes.4 Moreover, somatic mutations of the chemokine receptor CCR4 enhance ATL cell migration and cell growth, which can be countered by therapeutic anti-CCR4 antibodies.3,7 Although these earlier studies showed us how individual ATL players function, there was no unified understanding of how these players coordinate their actions to promote ATL. The study by Wong et al formally establishes the existence and functional importance of an IRF4-NF-κB network motif by which several players join forces to drive ATL cell proliferation and survival.
The article by Wong et al refers to an impressive array of chromatin profiling, gene expression, and functional studies in ATL cell lines and patient samples that confirms previous observations of high IRF4 expression in ATL and essential roles of IRF4, MYC, and NF-κB for ATL oncogene expression and cell viability. When Wong et al examined the patterns of where NF-κB and IRF4 bind the tumor cell DNA and regulate gene expression, they saw remarkable convergence far higher than what would be expected by chance. For example, they identified a highly expressed ATL gene that was cooperatively induced by NF-κB and IRF4 called baculoviral IAP repeat containing 3 (BIRC3). BIRC3 is an E3 ubiquitin ligase and member of the IAP family of proteins that inhibits apoptosis.8 Wong et al showed that BIRC3 was essential for ATL cell viability, establishing this protein as a novel ATL vulnerability. Besides BIRC3, additional nodes of convergence included CCR4, MYC, and several players in the T-cell receptor signaling pathway that activate NF-κB. Next, Wong et al delineated a key network connection in which NF-kB induced IRF4 expression, which is consistent with previous studies in normal T cells.9 This final link of the network established a pattern of regulation known in the jargon of mathematical modeling as a “type 1 coherent feed-forward loop”.10 In this pattern, transcription factor X induces transcription factor Y and then X and Y jointly induce gene Z (see figure). In all, Wong et al integrate new and old observations to reveal a unified perception of ATL oncogenesis consisting of viral inputs (Tax and HBZ), a propagating network motif (NF-kB and IRF4-BATF3), and oncogene outputs (MYC, CCR4, and BIRC3).
The Wong et al study was performed to stringent standards, resulting in high-quality signals, particularly for the chromatin profiling and gene expression data. Exceptional rigor was demonstrated by their use of multiple biological replicates, genetic tools, and cell lines that filled in gaps and confirmed and extended the literature. Even so, questions remain. Although the different ATL cell lines generally share the coherent feed-forward loop, it is unclear why there is heterogeneity. For example, the precise response elements that control MYC expression vary between cell lines, raising the possibility of additional players. In addition, mathematical simulations predict that the kinetics of the ATL feed-forward loop will be delayed when turning on and will be fast when shutting off. Although this feature seems attractive from a therapeutic standpoint, it is unclear how one can toggle the loop to the off state in the first place. Possibilities include small molecules that degrade key effectors or disrupt protein-protein interactions, the latter finding recent successes in breaking the menin-MLL interaction in myeloid leukemia. However, it remains unknown whether disrupting interactions between ATL players like IRF4 and BATF3 will show anti-leukemic effects or is even pharmacologically feasible.
In a remarkable and considerable undertaking, Wong et al stitched together several observations to define the satisfying architecture of a central ATL transcriptional network. We have learned that HTLV-1 is not only stealing the host cell’s machinery to make copies of itself and express viral oncoproteins, it is also rewiring the host cell’s transcriptional network to enhance its reproduction through cell division and subsequently ATL transformation. HTLV-1 evolved over thousands of years to hone its sinister plans to perfection. It is now up to us to find ways to expeditiously subvert the ATL network to treat this devastating disease.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal