

In this issue of Blood, identified NT5C2 thiopurine resistance mutations in 16% of children with relapsed B-cell acute lymphoblastic leukemia (B-ALL). Despite the fact that most of the mutations were subclonal, their presence was associated with an inferior outcome. Strikingly, NT5C2-mutated cells were sensitive to relapse therapy, suggesting a complex association between NT5C2 mutations and the poor prognosis of relapsed ALL.1
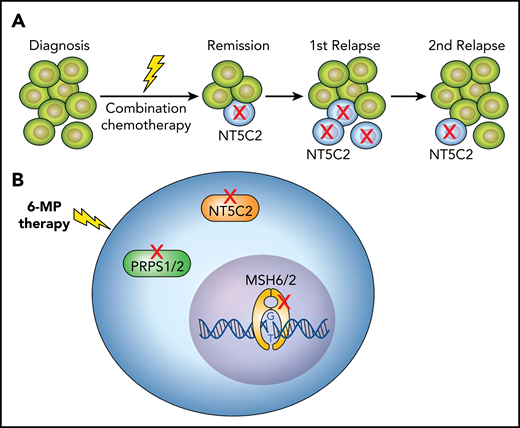
Chemotherapy-resistance mutations in relapsed ALL. (A) Most of the NT5C2 mutations identified in relapsed ALL are subclonal, identified in relatively few leukemic cells. In subsequent relapses, the subclonal NT5C2-mutated cells often disappeared or were diminished. (B) ALL relapse is frequently associated with different somatic mutations that confer thiopurine resistance, such as mutations in NT5C2 or PRPS1/2, which are involved in thiopurine metabolism, or mutations in MSH6/2, which induce thiopurine resistance via impaired DNA mismatch repair. 6-MP, 6-mercaptopurine.
Chemotherapy-resistance mutations in relapsed ALL. (A) Most of the NT5C2 mutations identified in relapsed ALL are subclonal, identified in relatively few leukemic cells. In subsequent relapses, the subclonal NT5C2-mutated cells often disappeared or were diminished. (B) ALL relapse is frequently associated with different somatic mutations that confer thiopurine resistance, such as mutations in NT5C2 or PRPS1/2, which are involved in thiopurine metabolism, or mutations in MSH6/2, which induce thiopurine resistance via impaired DNA mismatch repair. 6-MP, 6-mercaptopurine.
Leukemia may be compared with a small community in which all members (the cells) share the same founding mutations but also belong to distinct families (subclones) with unique genetic properties. This heterogeneity at the time of leukemia diagnosis2,3 is further enhanced at relapse.4,5 In humans, each family contributes to the livelihood of the village in its own way, but the role of subclones in promoting the survival and fitness of leukemia is unclear. This is one of the most fundamental questions in cancer biology, with far-reaching clinical implications. The article by Barz et al highlights both the clinical importance of subclones and the enigma regarding their biological role in leukemia propagation.
Leukemia relapse is caused by the expansion of cells that are resistant to chemotherapy because of preexisting or novel somatic mutations.6 Activating mutations in NT5C2, an enzyme that catabolizes thiopurines, have been identified in relapsed T- and B-ALLs (reviewed in Dieck and Ferrando7 ). Because mercaptopurine is a key element in ALL treatment and is administered for extended periods of time during maintenance therapy, the selection of NT5C2-mutated cells at relapse is not surprising. But if relapse itself was driven by NT5C2-mutated cells, one would expect all or most of the leukemic cells to carry these mutations.
Yet using highly sensitive polymerase chain reaction or next-generation sequencing, the authors showed that the majority of NT5C2 mutations, found in 16% of patients with relapsed B-ALL, were subclonal (ie, identified in relatively few leukemic cells) (see figure panel A). The mere existence of subclones of NT5C2-mutated leukemic cells was independently associated with a poor prognosis. Moreover, in subsequent relapses, the NT5C2-mutated cells often disappeared or were diminished. This is compatible with previous experimental findings by Tzoneva et al8 showing that leukemic cells carrying mutated NT5C2 have lower fitness, that is, impaired proliferative and self-renewal capacity. These observations suggest that NT5C2-mutated cells are not essential for the maintenance of relapsed leukemia, yet they predict poor outcome.
How does the presence of a subclone of NT5C2-mutated leukemic cells predict poor response to chemotherapy? What can we learn from these surprising observations regarding the possible role of subclones in maintaining the survival of the whole leukemia, the “community”?
One possible explanation is that the presence of an NT5C2 subclone is a surrogate marker for the coexistence of other subclones with chemotherapy-induced resistant mutations. This information is missing in the article by Barz et al, because the article does not include a comprehensive genomic analysis. An article recently published in Blood by Li et al6 demonstrated that approximately one quarter of all relapsed ALLs (similar to the 16% observed here) are characterized by therapy-induced mutations such as NT5C2. These alterations included other genes involved in resistance to thiopurines (mismatch repair genes: MSH2 and MSH6; thiopurine metabolism genes: PRPS1 and PRPS2; figure panel B), resistance to glucocorticoids (NR3C1, NR3C2), and resistance to methotrexate (FPGS), or resistance to DNA damaging agents such as P53. However, in the publication by Li et al, these mutations coexisted only rarely in the same leukemia.6 Thus, although it is likely that the presence of NT5C2 in a relapse subclone is a surrogate for a relapse-driving process, the nature of this process remains to be identified.
Another intriguing hypothesis to explain the association of subclonal NT5C2 mutations with poor prognosis relapse involves a non-cell-autonomous mechanism. Could NT5C2-mutated cells enhance the fitness and chemotherapy resistance of adjacent leukemic cells? This possibility has recently been experimentally demonstrated. FLT3-mutated subclones enhanced the fitness of experimental KMT2A-MLLT3 fusion leukemias by secreting the macrophage migration inhibitory growth factor.9 Is it possible that NT5C2-mutated cells in the bone marrow niche promote the survival and evolution of other leukemic subclones during remission of the primary ALL? Borrowing again from social sciences, this "collective impact" may be a general mechanism for coexistence or codependence of subclones that propagate leukemic cell resilience.
Beyond raising fascinating questions, the Barz et al study has 2 practical implications. First, molecular identification of NT5C2 may independently predict poor prognosis and may be used in risk stratification as an indicator for high-risk treatment. However, given the subclonal nature of NT5C2 mutations and their disappearance in subsequent relapses, specific therapy targeting the mutated NT5C2 cells8 at the time of relapse is unlikely to be beneficial. Whether targeting NT5C2-mutated cells during high-risk ALL first-line maintenance therapy will reduce the risk of relapse remains to be determined.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal