

CG Mullighan and T Haferlach: are co-senior authors
Introduction: Recent genomic sequencing studies have advanced our understanding of the pathogenesis of myeloid malignancies, including acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), and improved classification of specific subgroups. Unfortunately, these studies have mostly analyzed specific subtypes and/or used targeted DNA-sequencing, thus limiting discovery of novel mutational patterns and gene expression clusters. Here, we performed an integrated genome-wide mutational/transcriptomic analysis of a large cohort of adult AML and MDS samples to accurately define subtypes of diagnostic, prognostic and therapeutic relevance.
Methods: We performed unbiased whole genome (WGS) and transcriptome sequencing (RNA-seq) of 1,304 adult individuals (598 AML and 706 MDS; Fig. 1A), incorporating analysis of somatic and presumed germline sequence mutations, chimeric fusions and structural complex variations. Transcriptomic gene expression data were processed by a rigorous bootstrap procedure to define gene expression subgroups in an unsupervised manner. Associations between genetic variants, gene expression groups and outcome were examined.
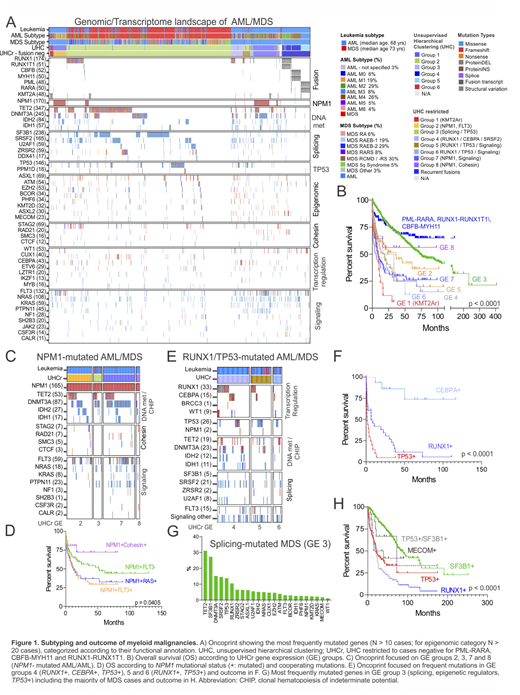
Results: Genomic/transcriptome sequencing confirmed diagnosis according to WHO 2016 of AML with recurrent genetic abnormalities in 10.9% of cases. These cases had a distinct gene expression profile (Fig. 1A), good prognosis (Fig. 1B) and a combination of mutations in the following genes: KIT, ZBTB7A, ASXL2, RAD21, CSF3R and DNM2 in RUNX1-RUNXT1 leukemia; FLT3, DDX54, WT1 and CALR in PML-RARA promyelocytic leukemia; KIT and BCORL1 in CBFB-rearranged leukemia. In addition, 9% of cases showed rearrangements of KMT2A, with known (e.g. MLLT3) and non-canonical partners (e.g. ACACA, and NCBP1) and poor outcome. Although common targets of mutations have been previously described for myeloid malignancies, the heterogeneity and complexity of mutational patterns, their expression signature and outcome here described are novel. Gene expression analysis identified groups of AML and/or MDS lacking recurrent cytogenetic abnormalities (87%). The spectrum of the most frequently mutated genes (>10 cases) and associated gene expression subtypes is summarized in Figure 1A. TET2 (more frequent in MDS than AML, p=0.0011) and DNMT3A (more frequent in AML than MDS, p<0.0001) were the most frequently mutated genes. Interestingly, mutations in these genes promoting clonal hematopoiesis were significantly enriched in the subgroup with NPM1 mutations. Overall, NPM1 mutations occurred in 27.4% of AML and 1% of MDS and were characterized by four expression signatures with different combination of cooperating mutations in cohesin and signaling genes and outcome (Fig. 1C, gene expression, GE, groups 2, 3, 7 and 8). Co-occurring NPM1 and FLT3 mutations conferred poorer outcome compared to only NPM1, in contrast co-occurring mutations with cohesin genes had better outcome (Fig. 1D). Additional mutations that significantly co-occurred with NPM1 were in PTPN11, IDH1/2, RAD21 and SMC1A. Three gene expression clusters accounted for additional 9% of cases with mutual exclusive mutations in RUNX1,TP53 and CEBPA and co-occurring with a combination of mutations in DNA methylation, splicing and signaling genes (Fig. 1E, GE groups 4, 5 and 6). Interestingly, RUNX1 mutations were significantly associated with SRSF2 mutations but not with SF3B1, showed high expression of MN1 and poor outcome (Fig. 1F). In contrast to the distinct, mutation-associated patterns of gene expression in AML samples, the gene expression profile of MDS was less variable despite diversity in patterns of mutation. MDS was enriched in mutations of SF3B1 (27.2%), mutually exclusive with SFRS2 (14.4%) and U2AF1 (5.5%); TP53 (13.7%) and RUNX1 (10.5%) and a combination of mutations in epigenetic regulators with outcome dependent on mutational pattern (Fig. 1A, G-H). Moreover, structural variations and/or missense mutations of MECOM accounted for 2% of cases.
Conclusions: the integration of mutational and expression data from a large cohort of adult pan myeloid leukemia cases enabled the definition of subtypes and constellations of mutations and have prognostic significance that transcends prior gene panel-based classification schema.
Meggendorfer:MLL Munich Leukemia Laboratory: Employment. Nadarajah:MLL Munich Leukemia Laboratory: Employment. Baer:MLL Munich Leukemia Laboratory: Employment. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Kern:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Mullighan:Illumina: Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: sponsored travel; Pfizer: Honoraria, Other: speaker, sponsored travel, Research Funding; AbbVie: Research Funding; Loxo Oncology: Research Funding; Amgen: Honoraria, Other: speaker, sponsored travel. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal