

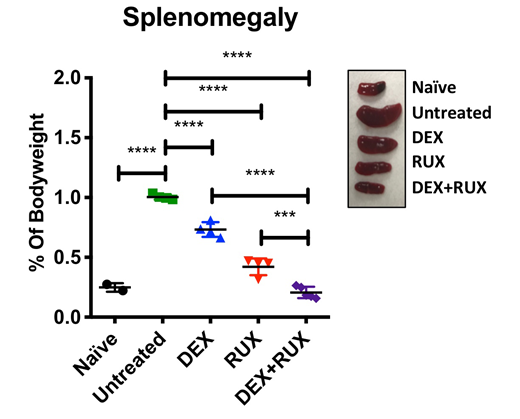
Hemophagocytic lymphohistiocytosis (HLH) is a severe immunologic syndrome characterized by a failure of CD8 T-cells to appropriately terminate immune responses, leading to excessive activation of immune cells that mediate life-threatening organ damage. Many patients with HLH are refractory to front-line therapy consisting primarily of the glucocorticoid dexamethasone (DEX) and the chemotherapeutic agent etoposide, resulting in five-year survival rates of only 62%. Therefore, novel strategies are needed to enhance the efficacy of frontline therapy in order to improve clinical outcomes. Hypercytokinemia is a hallmark feature of HLH due to the persistent activation of immune cells. CD8 T-cells both secrete and respond to these cytokines, making them important cellular targets of HLH therapy. Many HLH-associated cytokines activate the JAK/STAT pathway, and the JAK1/2 inhibitor ruxolitinib (RUX) has shown efficacy in murine models of HLH and in clinical case reports of refractory HLH. Importantly, JAK/STAT signaling has been shown in other disease contexts to induce DEX resistance. We therefore hypothesized that cytokine-mediated JAK/STAT signaling might contribute to DEX resistance in HLH and that this could be overcome by combination treatment with RUX. To test this hypothesis, we infected Prf1-/- mice with lymphocytic choriomeningitis virus (LCMV) to generate an in vivo model of primary HLH. Beginning on day four post-infection, mice were treated with vehicle control, DEX, RUX, or the combination of DEX and RUX and examined for signs of HLH. The Bliss independence model of synergy was applied to quantify the combinatorial effects of these drugs on disease parameters. Combined treatment with DEX and RUX synergistically lessened signs of systemic inflammation, including splenomegaly, numbers of inflammatory cells, including neutrophils and CD8 T-cells, and circulating levels of inflammatory cytokines. The mechanistic basis for these findings was then interrogated ex vivo. First, activated murine and human CD8 T-cells were exposed to DEX or etoposide in the presence of HLH-associated cytokines and were evaluated for cell viability. This revealed that the JAK-dependent cytokines IL-2 and IL-12 confer resistance specifically to DEX, but not to etoposide. IL-2 and IL-12 receptor signaling converge to activate STAT5. Consistent with this, exposure to RUX attenuated STAT5 activation in response to IL-2 or IL-12 stimulation. Furthermore, DEX and RUX synergized to induce cell death in the presence of IL-2 or IL-12. Mechanistic studies revealed that cytokine exposure did not inhibit nuclear translocation of ligand-activated glucocorticoid receptor (GR) or activation of GR transcriptional targets, suggesting that IL-2 and IL-12 act downstream of GR activity to confer DEX resistance. To interrogate this further, we quantified expression of BCL-2 family members in CD8 T-cells exposed to IL-2 or IL-12 and found significant upregulation of BCL-2 and BCL-XL. Using BH3 profiling, we functionally interrogated the intrinsic apoptotic pathway and found that cytokine exposure significantly suppressed the apoptotic potential of CD8 T-cells, such that DEX alone was no longer sufficient to induce apoptotic priming. However, concomitant exposure to RUX effectively restored apoptotic priming in response to DEX by inhibiting cytokine-induced upregulation of BCL-2 and BCL-XL. Finally, we performed BH3 profiling on cells from Prf1-/- mice following in vivo treatment with DEX +/- RUX. Similar to the ex vivo findings, LCMV infection and the subsequent onset of HLH significantly reduced the apoptotic potential of CD8 T-cells relative to cells from naïve mice, consistent with elevated levels of circulating cytokines in vivo. Combined treatment with DEX and RUX was more effective than either agent alone at inducing apoptotic priming, suggesting that both ex vivo and in vivo, cytokine exposure inhibits DEX-induced cell death by altering the cellular apoptotic potential. Taken together, this study reveals a mechanism of cytokine-mediated DEX resistance in HLH and provides rationale for combining DEX and RUX as a means to augment DEX sensitivity and improve clinical outcomes for patients with HLH.
Nichols:Incyte: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal