

Background:
Alpha-thalassemia (α-thalassemia) is an inherited hemolytic disease caused by complete or absent synthesis of α-globin chains due to alpha-globin chain synthesis disorders and is one of the most common genetic diseases in the world.
At present, the most commonly used strategies in the gene therapy research of thalassemia, the nonhomologous end joining (NHEJ) apparatus, is only suitable for the study of beta thalassemia and sickle-type anemia. The DNA double strand breaks have a high risk to induce frameshift mutations, leading to more serious clinical consequences. Therefore, it is still necessary to study a safer genetic repair program for alpha-thalassemia.
Methods:
The donor we chose to repair was from a patient with a genotype of -SEA/αCSα, whose mutation is one of the most common thalassemia alleles in Longyan city, and the HBA1 gene is normal. Due to the deletion of a copy of the HBA2 gene, only one copy of the mutation needs to be repaired, making it a suitable candidate for the study of alpha thalassemia gene therapy.
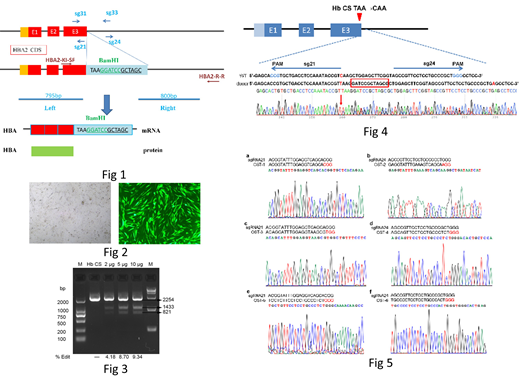
To minimize the risk of non-specific mutations introduced into the locus of interest, we used the single-base editing apparatus, dCas9 (dead Cas9), which only cleaves one strand of DNA and precisely modifies one base, effectively improve the specificity of target cleavage and reduce the risk of off-target. The recognition sequence on the homologous arm in the template was lengthened to reduce the risk of off-target.(see Figure 1 )
Result
We used the pmaxGFP plasmid as a positive control for optimizing electroporation conditions. More than 80% transfection efficiency was obtained (see Figure 2 )
Fig 2 pmaxGFP transfection to verify the transfection efficiency of fibroblasts ( 100x )
To investigate the effectiveness of sgRNA and donor plasmids for mutated gene repair, we analyzed the repair efficiency of fibroblasts. Primers were designed near the repair site and PCR sequences were amplified by PCR . After amplification of the genomic fragments, BamH1 digestion assay was performed.
Fig 3 PCR and Bam HI digested results using primers for the gene of interest, showed the gene editing efficiency was 4~10%.
The sequencing results showed that three clonals of α- thalassaemia mutations were successfully repaired.
Fig 4 Hb CS gene mutation target sequence repair and sequencing results
The sequencing results of Fig. 5 show that no mutation was detected at the six potential off-target sites.
Fig 5 off-target effect detection
The use of base editing is limited by off-target activity and DNA delivery efficiency to cells. Filtration into fibroblasts by plasmid electroporation, CRISPR/Cas9-mediated editing efficiency is usually 3~10%, and we achieve a gene editing efficiency of 4~10% (Figure 4).
Conclusions
Our data show that the gene transfer of normal HBA2 gene by electroporation the single-base editor CRISPR/dCas9 (D10A) and donor into cells can successfully repair Hb CS mutations.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal