

Background: Hematopoietic stem cell transplantation (HSCT) offers potentially curative therapy for patients with myelodysplastic syndrome (MDS) but disease progression after HSCT remains a major reason for failure after transplant. Identification of risk factors for progression of MDS after HSCT would allow to identify target population for early initiation of preventive treatments to improve outcomes.
Methods: Patients with a diagnosis of MDS who received first HSCT between 2013 and 2018 with available pre-transplant genetic profile obtained from next generation sequencing of genes were included for the retrospective analysis. Cytogenetic findings were categorized by the revised International Prognostic Scoring System (R-IPSS). Primary outcome of interest was risk of disease progression. Classification and regression tree (CART) analysis was performed to evaluate independent predictors on multivariate analysis using standard methods.
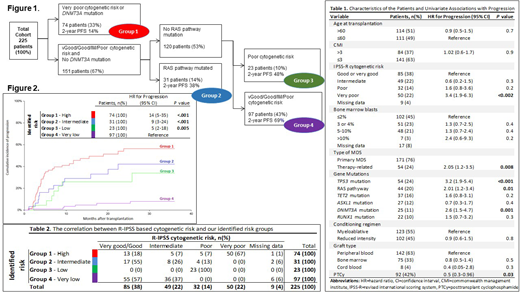
Results: Of 378 MDS patients transplanted within the study period, 225 were eligible to be included in this analyses. As shown in the table 1, the study cohort was high risk; cytogenetic risk groups were very-poor and poor in 50 (23%) and 32 (15%) patients, respectively. At least one pathogenic mutation was identified in 215 (91%) of patients prior to transplant. Most frequently mutated genes included, TP53 (24%, 54/225), RAS pathway genes (NRAS, KRAS, FLT3, PTPN11 and KIT) (20%, 44/225), TET2 (16%, 37/172), ASXL1 (12%, 27/172) and DNMT3A (11%, 25/200). In our cohort, patients with very-poor cytogenetics had a high frequency of TP53 mutations (73%), and TP53 mutations occurred almost exclusively in patients with very-poor cytogenetics (76% v 7%; P < .001). That makes those two groups almost inseparable from each other.
The median follow-up in 121 (54%) survivors was 24 months (range, 1.8 to 74 months). Of the 225 patients, 65 (29%) had disease progression after HSCT, with a median of 154 days to progression (range, 28 to 1196). By univariate analyses, presence of TP53 (HR, 3.2; CI, 1.9-5.4; P<.001), DNMT3A (HR, 2.6; CI, 1.5-4.7; P=.001), RAS pathway mutations (HR, 2.01; CI, 1.2-3.4; P=.01), therapy related MDS (HR, 2.05; CI, 1.2-3.5; P=.008), very poor risk cytogenetics (HR, 3.4; CI, 1.9-6.3; P<.002), and use of post-transplant cyclophosphamide (PTCy) (HR, 0.5; CI, 0.3-0.96; P=.003) were significant predictors of progression rate.
As previously mentioned, we used CART analysis to evaluate independent predictors of progression. The results demonstrated that given the significant overlap with TP53 and very poor cytogenetics, when both variables were forced into the model, only very poor cytogenetics remained significant for progression. Based on CART analysis, 4 mutually exclusive risk groups for progression were identified (Figure 1): high risk (very poor risk cytogenetics or DNMT3Amut), intermediate risk (good, intermediate or poor risk cytogenetics/RAS-pathmut/DNMT3Awt), low risk (poor risk cytogenetics/RAS-pathwt /DNMT3Awt) and a very low risk group (very good, good or intermediate risk cytogenetics/RAS-pathwt /DNMT3Awt). The correlation between R-IPSS based cytogenetic risk and our identified risk groups is shown in table 2. This illustrates how the addition of molecular data upstaged 25% of the patients to a higher risk category as well as downstaged 23% of the patients to a lower risk category for disease progression when compared to the original R-IPSS classification.
The cumulative incidence of disease progression at 2 years was 6% (reference), 26% (P=.005), 42% (P<.001) and 56% (P<.001) in very-low, low, intermediate and high risk groups, respectively (Figure 2). Within the risk groups identified, progression incidence was comparable by conditioning intensity and the use of PTCy.
The actuarial 2-year progression-free survival for the defined 4 risk groups was, 69% (reference), 48% (HR, 2; P=.04), 38% (HR, 2.2; P=.009), 22% (HR, 3.2; P<.001) and 14% (HR, 4.8; P<.001), in very-low, low, intermediate and high-risk groups, respectively. Non-relapse mortality was similar across the identified risk groups.
Conclusion: The proposed model, by incorporating DNMT3A and RAS pathway molecular mutation status to cytogenetic risk per R-IPSS, improves upon the classification of risk groups and enables the physician to better risk stratify and predict likelihood of progression after transplantation.
Garcia-Manero:Amphivena: Consultancy, Research Funding; Helsinn: Research Funding; Novartis: Research Funding; AbbVie: Research Funding; Celgene: Consultancy, Research Funding; Astex: Consultancy, Research Funding; Onconova: Research Funding; H3 Biomedicine: Research Funding; Merck: Research Funding. Popat:Bayer: Research Funding; Incyte: Research Funding; Jazz: Consultancy. Ciurea:Kiadis Pharma: Membership on an entity's Board of Directors or advisory committees, Other: stock holder; Spectrum: Membership on an entity's Board of Directors or advisory committees; Miltenyi: Research Funding; MolMed: Membership on an entity's Board of Directors or advisory committees. Kebriaei:Jazz: Consultancy; Pfizer: Honoraria; Kite: Honoraria; Amgen: Research Funding. Bashir:Imbrium: Membership on an entity's Board of Directors or advisory committees; Amgen: Membership on an entity's Board of Directors or advisory committees; Spectrum: Membership on an entity's Board of Directors or advisory committees; Kite: Membership on an entity's Board of Directors or advisory committees; Takeda: Membership on an entity's Board of Directors or advisory committees, Research Funding; StemLine: Research Funding; Acrotech: Research Funding; Celgene: Research Funding. Champlin:Actinium: Consultancy; Johnson and Johnson: Consultancy; Sanofi-Genzyme: Research Funding. Oran:Astex pharmaceuticals: Research Funding; AROG pharmaceuticals: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal