

Background
Bone disease (BD) is a hallmark of multiple myeloma (MM) and is characterized by severe skeleton damage, reduced quality of life and overall survival (1-2). Several findings indicated that IL-17 producing CD4+ T cells (Th17) play a central role in triggering MMBD and support MM cell growth mainly by IL-17 production. There is compelling evidence that miR-21 is a central player in Th17 effector functions. Our preliminary data have shown that miR-21 is highly upregulated in MM-Th17 isolated from patients with active BD as compared to MM with no active BD and controls. We found that inhibition of miR-21 in naive T cells (miR-21i-T cells) impaired differentiation towards Th17 in vitro, by reducing interleukin (IL)-17, IL-22, RANKL and RORC, leading to abrogation of osteoclast (OCL) bone resorption.
Aims
Based on these premises, we sought to explore miR-21 related underlying molecular networks that support pathogenic Th17 differentiation and function. As miRNAs may exert direct and indirect effects on gene expression and at post-transcriptional level, we performed a global head-to-head comparison by RNA-seq and proteomic -phosphoproteomic analysis on miR-21i-Th17. Then, we recapitulated and validated our findings in NOD/SCID gNULL mice, injected intratibially with miR-21i-T cells and MM cells.
Methods
RNAseq and proteomic/phosphoproteomic assays have been performed on in vitro differentiated Th17 cells originated from scramble control (SC) or miR-21i transfected naïve T cells (SC-Th17 and miR-21i-Th17 respectively) from 3 healthy donors through MARS-seq protocol adapted for bulk RNA and proteome/phosphoproteome analysis . Data have been analyzed through R by using different packages including limma, DESEQ2 and pheatmap. To perfom global proteome/phosphoproteome analysis, we conducted a mass spectrometry study of phosphopeptides protein extract from SC-Th17 and miR-21i-Th17, enriched using SCX-IMAC/TiO2. High-resolution LC-Ms/MS data were processed using Proteome Discoverer software
Results
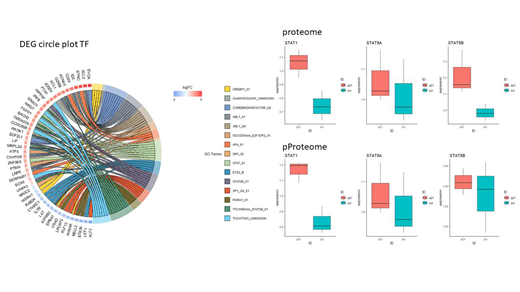
In the presence of miR-21i, we found 109 upregulated and 22 downregulated proteins in the global proteome analysis of Th17 cells, while 90 and 18 phosphoproteins were up and down modulated, respectively. Paired analysis showed that 46 proteins are modulated in expression but not in phosphorylation, 23 proteins are modulated in phosphorylation but not in expression, while 85 proteins are modulated in both conditions. These data suggest that selective miRNA modulation interferes with a specific and limited group of proteins/phosphoproteins according to cell type and despite predicted pleiotropic miRNA activity. To understand whether miR-21i-Th17 undergo a "molecular reprogramming", we evaluated gene expression by RNA seq Analysis of miR-21-related molecular pathways in Th17 cells and found upregulation of STAT-1/-5a-5b, downregulation of STAT-3 and redirection of Th17 to Th1/activated like cells as shown by a pair-to-pair RNAseq and proteome/phosphoproteome analysis. These data indicate that miR-21 plays a central role in driving Th17 differentiation and function in a proinflammatory milieu such as MM-Bone marrow microenvironment (BMM). However, when miR-21 activity is strongly counteracted, pathogenic Th17 can switch to a Th1 like phenotype (STAT 1 dependent gene/protein upregulation). This switch may partly explain the attenuation of MMBD observed in vitro. To confirm our observation in vivo, we injected intratibially miR-21i exposed- or scramble miR (SC) exposed-naïve CD4+ T cells together with MM cells into gamma null SCID mice. We observed that mice injected with SC CD4+ naïve T cells presented severe local skeleton damage, while bone structure was preserved in miR-21i naïve CD4+ T cells injected mice.
Conclusions
Our data highlight the relevance of miR-21 in supporting Th17 mediated MMBD onset and progression. The possibility to "reprogram" MM Th17 by miR-21 modulation opens a new avenue to develop miR-21 targeting therapeutic strategies to counteract BMM-dependent MM development and related-BD.
Paiva:Amgen, Bristol-Myers Squibb, Celgene, Janssen, Merck, Novartis, Roche, and Sanofi; unrestricted grants from Celgene, EngMab, Sanofi, and Takeda; and consultancy for Celgene, Janssen, and Sanofi: Consultancy, Honoraria, Research Funding, Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal