

Introduction: Aberrant cell-cell interactions involving the endothelium are central to the pathophysiology of crises in sickle cell disease (SCD), including acute chest syndrome (ACS). We previously demonstrated that the plasma of SCD patients contains circulating small extracellular vesicles (EVs) and that those vesicles can disrupt endothelial integrity in vitro, including a decrease in VE-cadherin. The current study was designed to examine the effects of those EVs on additional components of the endothelial junctions including tight (zonula occludens 1, ZO-1) and gap junctions (connexin43, Cx43) and to test the hypothesis that the junctions would be more severely affected by EVs isolated from patients during an episode of ACS than by those isolated from the same patient at baseline.
Methods: We identified subjects with SCD in our biobank who had plasma isolated at baseline and at the beginning of an admission for ACS (prior to transfusion). Samples were considered baseline if the patient was more than 4 weeks since transfusion and had no new health-related complaints. ACS was defined by the presence of an infiltrate on chest x-ray combined with fever, pain, hypoxia or cough. EVs were isolated from plasma using established methodologies. To determine the effects on endothelium, cultures of human microvascular endothelial cells were treated with EVs for 48 h. Cells were fixed and studied by fluorescence microscopy (after immunolocalization of Cx43, ZO-1 and/or VE-cadherin and staining of nuclei with DAPI). Proteins were detected and quantified by immunoblotting. mRNA expression was determined by RT-qPCR. Gap junction mediated intercellular communication was assessed following microinjection of Lucifer yellow and neurobiotin.
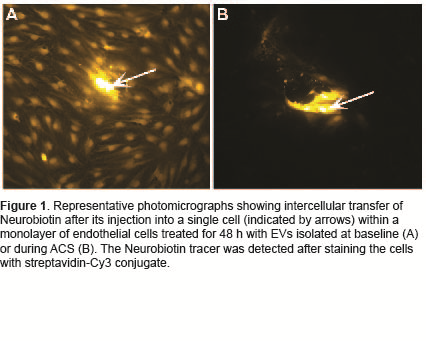
Results: Microscopy confirmed our previous observation that EVs isolated from subjects with SCD caused in vitro disruption of endothelial monolayers and that damage is significantly worse when EVs are isolated during an episode of ACS. The distribution and abundance of VE-cadherin and ZO-1 at the plasma membrane of undisturbed cells were minimally affected by SCD EVs. While baseline EVs did not detectably affect the distribution of Cx43, EVs isolated during ACS caused a loss of Cx43 from the plasma membrane. The integrated intensity of Cx43 membrane staining was decreased by ~20% following treatment with ACS EVs. Cx43 protein decreased on average by 32 % and Cx43 mRNA levels by 21% in cells treated with ACS EVs compared to baseline from the same patient. EVs isolated during ACS caused significant disruption in intercellular transfer compared to EVs isolated at baseline (67-94% reduction) (Figure 1).
Conclusions: Our results show that subjects with SCD produce small EVs that cause disruption of the endothelial monolayer in vitro. Gap junctions composed of Cx43 are the most sensitive of the cell-cell junctions in this setting, since their abundance and function are reduced by ACS EVs even when the endothelial monolayer appears intact. Disruption of endothelial intercellular communication mediated by Cx43 appears to be an early and sensitive event in the endothelial disturbance caused by EVs in SCD patients.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal