

RNA splicing is a fundamental biological process that generates protein diversity from a finite set of genes. Recurrent somatic mutations of genes involved in RNA splicing are present at high frequency in Myelodysplasia (up to 70%) but less so in Acute Myeloid Leukemia (AML; less than 20%). To investigate whether there were aberrant and recurrent RNA splicing events in the AML transcriptome that were associated with poor prognosis in the absence of splicing factor mutations, we developed a bioinformatics pipeline to systematically annotate and quantify alternative splicing events from RNA-sequencing data (Fig A).
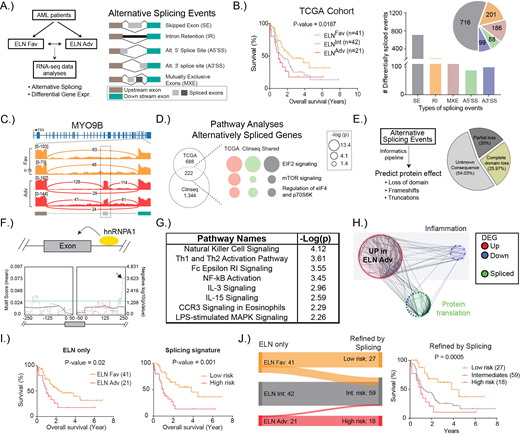
We first analysed publicly available RNA-seq data from The Cancer Genome Atlas (TCGA, n=170). We focussed on non-M3 AML patients with no splicing factor mutations (based on reported genomic sequencing and verified by re-analysis of RNA-seq data from all patients) who had received intensive chemotherapy. We segregated these patients based on their European Leukaemia Net (ELN) risk classification and identified 1290 alternatively spliced events impacting 910 genes that were significantly different (FDR<0.05) between all ELNAdv (n=41) versus all ELNFav patients (n=21, Fig B). The majority were exon skipping events (716 events, 62%, Fig B-C), followed by intron retention (201 events, 15.6%, Fig B). We next used RNA-seq data from a second non-M3 AML patient cohort (ClinSeq- Sweden; ELNAdv, n=75 and ELNFav, n=47), detecting 2507 events mapping to 1566 genes. Comparing across the two cohorts, 222 shared genes were detected to be affected by alternative splicing (Fig D). Ingenuity pathway analysis associated these genes with pathways related to protein translation.
In order to prioritise those alternatively spliced events most likely to have a deleterious function, we developed an analytical framework to predict their impact on protein structure (Fig E). 87 alternatively spliced events, 25.81% of the commonly shared splicing events, relating to 78 genes (35.13% of all genes) were predicted to directly alter highly conserved protein domains within the affected genes, leading to either a complete (~25%, Fig E) or a partial loss of a domain (20%, Fig E). These in silico predictions are likely to be an underestimate of the true impact, as splicing alterations mapping to poorly annotated domains or affecting the tertiary structure of proteins would be missed. A number of splicing factors themselves were differentially spliced, with the alternative splicing predicted to have functional consequences. This was exemplified by hnRNPA1, a factor with well-established roles in splicing, is itself alternatively spliced in patients and predicted to be deleterious. Consistent with this, motif scanning analyses indicated that a number of mis-spliced transcripts had hnRNPA1 binding motifs (Fig F).
To assess the impact of these alternatively spliced events (that were predicted to also disrupt highly conserved protein domains) on the transcriptome, we simultaneously quantified differential gene expression. IPA analysis of the 602 genes that were differentially expressed between ELNAdv and ELNFav patients and shared between both TCGA and ClinSeq cohorts indicated that they were associated with pathways (Fig G) that were distinct from those associated with aberrantly spliced genes (Fig D). A number of pathways related to inflammation were enriched amongst the genes observed to be upregulated in ELNAdv patients (Fig G). Network analyses integrating the alternatively spliced genes with differentially expressed genes revealed strong interactions (Fig H), indicating functional associations between these biological events.
Given these strong network interactions, we investigated the potential prognostic significance of these alternatively spliced events. To this end, we utilised machine-learning methods to derive a "splicing signature" of four mis-spliced genes with a predictive capacity equivalent to the ELN (Fig I). The splicing signature further refined existing risk prediction algorithms to improve the classification of patients (Fig J). Taken together, we report the presence of extensive deregulation of RNA splicing in AML patients even in the absence of splicing factor mutations. Many of these events were shared in patients with adverse outcomes and their impact on the AML transcriptome points towards vulnerabilities that could be targeted.
Unnikrishnan:Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding. Lehmann:TEVA: Consultancy, Membership on an entity's Board of Directors or advisory committees; Pfizer: Membership on an entity's Board of Directors or advisory committees; Abbive: Membership on an entity's Board of Directors or advisory committees. Pimanda:Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal