

Introduction: Patients who develop therapy-related myeloid neoplasms (t-MN) have dismal outcomes. Previous studies reported the incidence and risk factors associated with t-MN development. Lenalidomide, in the setting of oral, but not intravenous, melphalan is associated with a higher risk of t-MN (Palumbo et. al, Lancet Oncology, 2014). We carried out this study to evaluate the clinical and pathologic features of t-MN, therapies employed, and factors that predict long-term survival after diagnosis.
Patients and methods: We identified patients who received the first ASCT 1998-2016 at our institution. t-MN was defined per the WHO 2016 classification. Median overall survival (OS) was calculated from the time of t-MN diagnosis to last follow-up or death. Statistical analyses were performed using SAS (JMP v14.1) or GraphPad Prism (v7).
Results: Out of 2115 patients that underwent at least one ASCT, 53 (2.5%) developed t-MN. Thirty-five of 53 (66%) patients who developed t-MN had received lenalidomide. Among 2062 patients that did not develop t-MN, 916 (44.4%) patients received lenalidomide. Lenalidomide exposure was associated with development of t-MN (χ2 with Yate's correction 8.9, p=0.003).
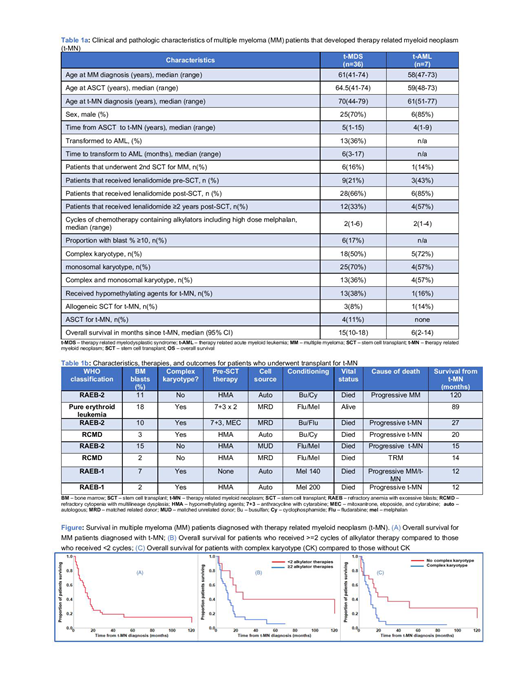
Ten patients were excluded from further analyses due to lack of follow up. Clinical characteristics are shown in Table 1a (N=43). Median age at t-MN diagnosis was 70 years (range 44-79). Median time from ASCT to t-MN was 5 years (range 1-15). After a median follow-up of 70 months (95% CI, 38-134), the median OS was 12 months (95% CI, 9-17, Figure). Primary causes of death were t-MN (71%), MM (12%), both (6%), and other including infection, GVHD, and unknown (12%). Seven (16%) had t-AML and 36(84%) had t-MDS. Three (42%) of 7 patients with t-AML had pure erythroid phenotype. At the time of last follow-up, 9 (21%) were alive. Seven (17%) underwent two ASCT, 16 (36%) received more than 2 years cumulative dose of lenalidomide. Median number of cycles of alkylator therapy including high-dose melphalan (HDM) used for ASCT was 2 (range 1-6).
On univariate analysis, factors predicting OS from t-MN diagnosis were ≥ 2 alkylator vs. < 2 cycles (11 vs. 27 months, p=0.02), ≥10% vs. <10% blast at the time of t-MN diagnosis (5.5 vs. 17 months, p=0.01), and the presence of complex karyotype (CK) vs. not (11 vs. 17 months, p=0.03). There was no difference in survival among t-MDS patients who transformed to t-AML vs. those who did not, those who received 1 vs. 2 ASCT, and those with lenalidomide duration ≥ 2 vs. <2 years. On multivariate analysis, the number of alkylator therapies and CK, but not % blasts predicted OS from t-MN diagnosis (Figure).
Eight (19%) patients proceeded to receive an autologous (n=4) or allogeneic (n=4) transplant for t-MN (Table 1b). There was no survival advantage for patients who underwent SCT for t-MN, compared to those who did not (17 vs. 10 months, p=0.3). Similarly, there was no survival advantage for patients who received allo SCT compared to auto SCT (21 vs. 16 months, p=0.7). Survival for t-MDS and t-AML was also not different (15 vs. 6 months, p=0.2). There is only one long-term survivor in the transplant group (s/p allogeneic SCT). Four of seven (57%) patients died of progressive t-MN, 1 died of progressive MM, progressive MM and t-MN, and TRM each.
Conclusions: Lenalidomide, in the setting of HDM, is associated with an increased risk of t-MN. Increased exposure to alkylators, a higher tumor burden, and the presence of CK predict poor survival in t-MN. As HDM/ASCT followed by lenalidomide maintenance remains the standard of care for eligible patients, minimizing exposure to other alkylator regimens may be prudent. While considered the standard, only a minority undergo transplant; and post-transplant outcomes remain poor. Studies to identify prognostic genetic and epigenetic risk factors are ongoing.
Al-Kali:Astex Pharmaceuticals, Inc.: Research Funding. Kumar:Celgene: Consultancy, Research Funding; Janssen: Consultancy, Research Funding; Takeda: Research Funding. Patnaik:Stem Line Pharmaceuticals.: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal