

Myelodysplastic syndromes (MDS) are a heterogeneous group of hematopoietic stem cell disorders for which allogeneic hematopoietic stem cell transplantation (HCT) is currently the only curative treatment. Epigenetic lesions are considered a major pathogenetic determinant in many cancers, including MDS, and combination epigenetic therapies have emerged. In this study, we hypothesize that interplay between key epigenomic signatures in the MDS patient undergoing HCT and their donor epigenomic profile serve as a prognostic factor of post-HCT MDS relapse risk.
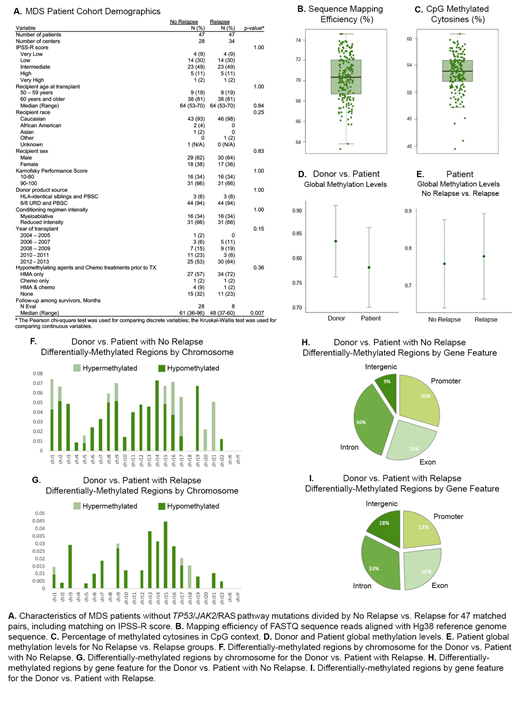
Reduced representation bisulfite sequencing (RRBS) was chosen to identify genome-wide epigenetic alterations as a cost-efficient method for building large data resources that reduces sequence redundancy and selects only CpG-rich regions of the genome for sequencing. A unique cohort of 188 samples from the Center for International Blood and Marrow Transplant Research (CIBMTR) biorepository was sequenced through RRBS. This cohort was composed of 94 pre-transplant samples from MDS patients that received peripheral blood stem cell grafts and were selected as case/controls for post-HCT relapse/non-relapse matched on patient, disease and transplant characteristics. The remaining 94 samples were from the patients' healthy allogeneic donors. Only patient samples that were wild-type for previously-identified MDS-prognostic TP53, RAS pathway and JAK2 mutations were included in this cohort to promote discovery of novel factors. We developed methylPrep, a Python application, to filter the low methylation calls and group shared sites by donors, relapsed patients and non-relapsed patients.
We comprehensively identified differentially methylated regions (DMRs) by comparing the methylation patterns in healthy donors and MDS patients that relapsed or did not relapse. The healthy donor group displayed higher global methylation levels (GML) than the patient group as a whole, and the relapsed patient showed higher GML than the non-relapsed patient, though these differences were not statistically significant, and we continue to investigate whether hypo-methylating agents play a role. We selected high DMRs (50-bp interval), with at least 25% difference in methylation calls, using Fisher's exact test, where the threshold q-value equals 0.05, and uncovered 367 significant hyper-DMRs and 38 significant hypo-DMRs in donors compared to patients genome-wide. For disease relapsed versus non-relapsed MDS patients, we identified 121 hyper-DMRs and 64 hypo-DMRs, and the distribution of DMRs was highly varied. Furthermore, we compared epigenome compatibility between donors and patients who did or did not relapse after transplantation and discovered a distinct difference in DMR patterns from chromosome to chromosome and through region annotation. Interestingly, a higher number of DMRs were located in promoter regions between donors and non-relapsed patients versus donors and disease-relapsed patients.
Identified DMRs, especially those located in promoter regions, may be involved in regulation of gene expression. These promoter DMRs may serve as candidate indicators or sites for potential diagnosis and therapy selection for MDS patients and may aid in the prediction of transplant outcomes and matching of the best donor for the MDS patient. Continued investigation will enable validation and assessment of the impact and mode of action for these distinct methylation signatures and global methylation patterns in MDS associated with HCT outcomes.
Nazha:Incyte: Speakers Bureau; Daiichi Sankyo: Consultancy; Jazz Pharmacutical: Research Funding; Novartis: Speakers Bureau; Tolero, Karyopharma: Honoraria; MEI: Other: Data monitoring Committee; Abbvie: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal