

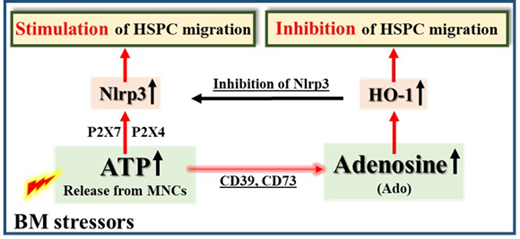
Background. In order to develop more efficient mobilization strategies, we have to better understand the mobilization process at the molecular and cellular levels. We envision pharmacological mobilization of HSPCs as the result of "sterile inflammation" in the bone marrow (BM) microenvironment induced by pro-mobilizing agents (e.g., G-CSF or AMD3100). This leads to activation of the complement cascade (ComC), which is required for egress of HSPCs from BM into peripheral blood (PB) (Leukemia 2018; 32:1116-1123). In support of this hypothesis, we reported recently that BM-residing innate immunity cells (e.g., monocytes, granulocytes and dendritic cells) release adenosine triphosphate (ATP) into the extracellular space during the initiation phase of mobilization. ATP released from the cells is an important signaling molecule that activates purinergic signaling receptors on the surface of hematopoietic cells (Leukemia 2018, 32:1920-1931) and is released mainly by pannexin 1 channels. Inhibition of this channel significantly decreases mobilization efficacy (Leukemia 2018, 32:1920-1931). In addition, ATP in the extracellular BM space is processed by the CD39 and CD73 ectonucleotidases to adenosine (Ado), which inhibits mobilization. To shed more light on the molecular mechanism governing mobilization of HSPCs, we focused on the role of the Nlrp3 inflammasome, which is highly expressed in hematopoietic cells. Moreover, while ATP binds to the P2X7 and P2X4 purinergic receptors and strongly activates Nlrp3, Ado inhibits migration of HSPCs in a heme oxygenase 1 (HO-1)-dependent manner.Hypothesis. We hypothesized that the Nlrp3 inflammasome is a link or "cogwheel" between purinergic signaling and the ComC that orchestrates optimal mobilization of HSPCs. Materials and Methods. To test this hypothesis we first mobilized wild type mice by G-CSF and AMD3100 administration in the presence of the small-molecule Nlrp3 inhibitor MCC950 or the Nlrp3 inflammasome activator nigericin, and the results were subsequently compared with results for Nlrp3-KO mice. Following mobilization, we measured i) the total number of white blood cells (WBCs) and ii) the number of circulating clonogenic colony-forming unit granulocyte/macrophage (CFU-GM) progenitors and Sca-1+c-kit+lineage- (SKL) cells circulating in PB. The secretion of ATP from BM cells was inhibited by employing the pannexin-1-blocking drug probenecid or a synthetic pannexin-1-blocking peptide (Panx-1). Nlrp3 inflammasome mRNA expression was measured in BM innate immunity cells, CD11b/Gr-1+ cells, and CD11b/Gr-1+ cells isolated under steady-state conditions from mobilized mice stimulated with ATP or Ado. Elements of the activated inflammasome, such as caspase-1, IL-1b, and IL-18, were measured at the mRNA (RT-PCR) and protein (ELISA) levels. In parallel, we evaluated i) the expression of HO-1 in HSPCs by employing RQ-PCR and western blot analysis and ii) activation of the ComC by C5a ELISA. Results. While mobilization of HSPCs was significantly decreased in P2X7-KO mice and mice with Nlrp3 inflammasome deficiency, it was significantly increased in the presence of the Nlrp3 inflammasome activator nigericin. Proper activation of the inflammasome required ATP release in a pannexin-1-dependent manner and stimulation of hematopoietic cells in a P2X7 purinergic receptor-dependent manner. Moreover, Ado inhibited the mobilization process by upregulating HO-1 in HSPCs, which, as we observed, inhibits the Nlrp3 inflammasome in HSPCs. Finally, while activation of the Nlrp3 inflammasome was positively correlated with activation of the ComC, inhibition of the Nlrp3 inflammasome had the opposite effect. Conclusions. We demonstrate for the first time that purinergic signaling involving ATP and its metabolite adenosine regulate the mobilization of HSPCs in an Nlrp3 inflammasome-dependent manner. While ATP triggers and promotes this process via activation of the Nlrp3 inflammasome, adenosine has an inhibitory effect by upregulating HO-1 (Figure 1). Based on these findings, stimulation of the Nlrp3 inflammasome by ATP, inhibiting the Ado level by small-molecule inhibitors of the CD39 or CD73 enzymes involved in generation of extracellular Ado, or direct inhibition of HO-1 in HSPCs by employing small-molecule inhibitors may provide the basis for more efficient mobilization strategies.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal