

Introduction: Multiple myeloma (MM) is a malignancy of antibody-secreting plasma cells (PCs) characterized by the clonal expansion and accumulation of monotypic PCs in the bone marrow (BM). Despite the remarkable improvements have been done in knowledge on the pathobiology of MM, therapy and medical care, MM still remains an incurable disease. Therefore elucidation of regulatory circuits altered such as long non-coding RNAs (lncRNAs) will allow us to obtain a better understanding into the biology and targeted therapy of MM. Overwhelming evidence suggests that lncRNAs have huge impact on adjusting gene expression through the processes of transcription and post-transcription regulation, genomic imprinting, and chromatin modification. In our previous study, we validated one lncRNA named ST3 beta-galactoside alpha-2,3-sialyltransferase 6 antisense RNA 1 (ST3GAL6-AS1) which was upregulated markedly in MM patients. Further research revealed that ST3GAL6-AS1 promote the adhesion, migration and invasion ability of MM cells. To determine the precise mechanism underlying ST3GAL6-AS1 biological function, we expound its interactive protein in this study.
Methods: Comprehensive Identification of RNA binding Proteins by Mass Spectrometry (ChIRP-MS) was utilized to identify interactive protein of ST3GAL6-AS1. MM cell line MM.1S was reversibly crosslinked with UV/formaldehyde under native condition, with or without prior lysis, then hybridized with biotinylated DNA antisense and pulled down with streptavidin dynabeads. Peptides were identified by MS eventually. Raw MS files were processed with MaxQuant (Version 1.5.6.0). Both peptide and protein FDR should be less than 0.01. Gene Ontology (GO) analysis (http://david.abcc.ncifcrf.gov/summary.jsp) was used to analyze differentially expressed transcripts. Pathway analysis of differentially expressed transcripts was accomplished using Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg). String online software (http://www.string-db.org/) was utilized to predict protein-protein interactions.
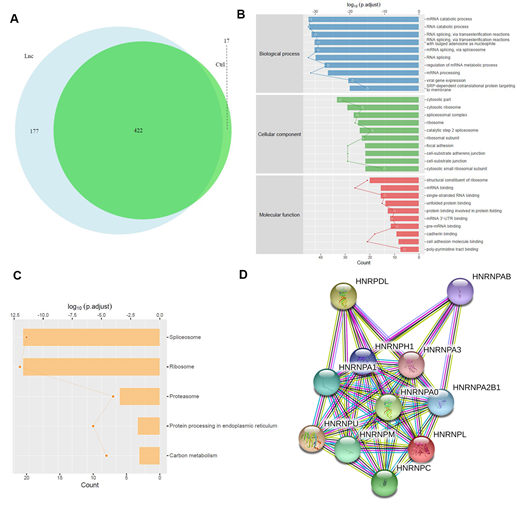
Results: ChIRP-MS analysis identified a total of 599 proteins were interacted with ST3GAL6-AS1, among them, 177 proteins were identified to bind with ST3GAL6-AS1 specifically compared with control group (Figure 1A). GO and KEGG analysis were based on these 177 specific proteins. GO analysis showed that the top ten enriched biological process (BP) were mainly involved in mRNA catabolic process and splicing, while cellular component (CC) was cytosolic part and molecular function (MF) was structural constituent of ribosome (Figure 1B). KEGG pathway analysis of the top five enriched pathways included spliceosome and ribosome (Figure 1C). In the specific interaction proteins, we found multiple proteins belong to heterogeneous nuclear ribonucleoproteins (hnRNP) family, such as hnRNPA, hnRNPU. Further research showed that involved hnRNPs interacted with each other (Figure 1D) and mainly took important role in spliceosome by GO and KEGG analysis (FDR<0.01). The results above suggested that ST3GAL6-AS1 might regulate target mRNA splice and catabolism through its interactive proteins, especially hnRNP family.
Conclusions: We have identified a group of proteins in MM cells which were specifically interacted with lncRNA ST3GAL6-AS1. These proteins mainly participate in mRNA catabolism and spliceosome, which might was the mechanism of ST3GAL6-AS1 to regulate its target gene in MM. Further investigation will focus on the exact action of ST3GAL6-AS1 through its interacting proteins.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal