

Defibrotide is a mixture of single-stranded phosphodiester oligonucleotides 9-80 bases in length derived from mammalian tissue and it is the only medication that is FDA approved for the prevention and treatment of post-transplant veno-occlusive disease, also known as sinusoidal obstructive syndrome (VOD/SOS). The mechanism of action of defibrotide is not well understood. A better understanding of the molecular mechanism by which defibrotide prevents and reverses the microvascular occlusion seen in VOD/SOS will be critical in the development of novel therapeutics for VOD/SOS and other related disorders.
Our central hypothesis is that single stranded phosphodiester oligonucleotide mixtures such as defibrotide alter thrombin-induced platelet and endothelial cell activation primarily via altered signaling through protease activated receptors (PARs).
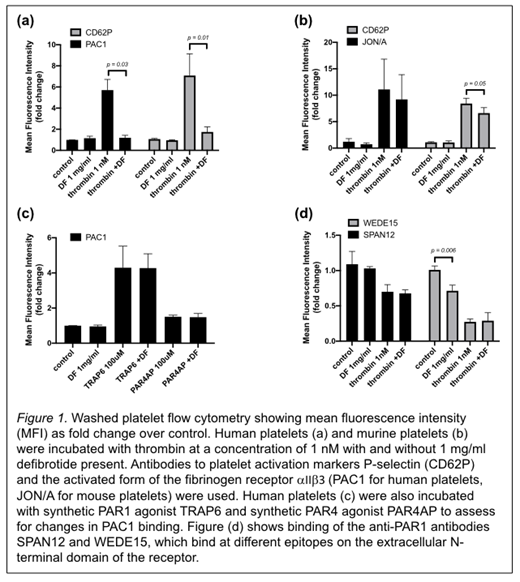
To determine the effect of defibrotide on thrombin-induced platelet activation we performed flow cytometry on washed platelets from humans and mice. Washed platelets were incubated with agonist (1 nM thrombin, 100 μM synthetic PAR1 agonist TRAP6, or 100 μM synthetic PAR4 agonist PAR4AP) in the presence or absence of 1 mg/mL of defibrotide. Fluorescent antibodies against P-selectin (anti-CD62P antibody) and against the high affinity conformation of the αIIβ3 receptor (PAC1 antibody for human samples and JON/A antibody for murine samples) were added to assess for inhibition of platelet activation by defibrotide. Fluorescent anti-PAR1 antibodies SPAN12 (which spans the canonical thrombin cleavage site) and WEDE15 (which binds at the hirudin-like domain) were also added to separate samples to assess whether defibrotide inhibits cleavage of the PAR1 N-terminal domain by thrombin. After twenty minutes of incubation at room temperature the samples were analyzed by flow cytometry. We calculated the geometric mean of the fluorescence intensity (MFI) for three replicate samples and data are shown as the fold change compared to the washed platelet control without addition of agonist or defibrotide.
For thrombin-stimulated human platelets, both anti-CD62P binding (MFI fold over control 5.7 vs. 1.2) and PAC1 antibody binding (MFI fold over control 7.1 vs. 1.7) are significantly reduced in the presence of defibrotide (See Figure 1a). For thrombin-stimulated murine platelets there is a small decrease in anti-CD62P antibody binding (MFI fold over control 8.4 vs. 6.6), but no significant change in JON/A antibody binding (See Figure 1b). Binding of both anti-CD62P and PAC1 antibodies to human platelets stimulated with synthetic PAR agonists was unaffected by the presence of defibrotide (See Figure 1c). There is no significant difference in SPAN12 or WEDE15 binding in thrombin-stimulated platelets in the absence or presence of defibrotide. However, defibrotide alone does appear to interfere with WEDE15 binding in unstimulated platelets (See Figure 1d).
In conclusion, we showed that defibrotide inhibits thrombin-induced human platelet activation at low concentrations of thrombin, as shown by a decrease in surface platelet activation markers measured by flow cytometry. This effect is much less pronounced under the same conditions if mouse platelets are used, supporting the hypothesis that defibrotide inhibits platelet activation by alteration of PAR1 receptor signaling, since PAR1 receptors are not present on mouse platelets. Defibrotide does not interfere with TRAP6-induced platelet activation, evidence against interference with the intracellular portion of the signaling pathway, since TRAP6 is a synthetic PAR1 agonist that binds directly into the ligand-binding pocket of the receptor, eliminating the need for cleavage of the PAR1 extracellular domain. The decrease in WEDE15 antibody binding to washed platelets seen with defibrotide alone suggests that the mechanism may involve alterations in the hirudin-like domain of the PAR1 receptor which is crucial for thrombin binding and cleavage site specificity. Ongoing studies including investigating the effect of defibrotide on peptide mimics of the extracellular N-terminal domain of the PAR1 receptor and the effect of defibrotide on the activation of endothelial cells will likely shed light on the exact mechanism of platelet response modulation by defibrotide and may provide new targets for treatment of the microvascular occlusion that characterizes VOD/SOS.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal