

BACKGROUND: Chronic red blood cell (RBC) hemolysis that accompanies sickle cell disease (SCD) is associated with elevated iron storage in tissues, in particular the liver (Nguyen et al. 2014). Iron storage within the liver leads to oxidative stress and fibrosis, reducing circulating antioxidants (vitamin D & hemopexin) and essential hormones, namely insulin-like growth factor-1 (IGF-1). We have previously shown that SCD mice progressively develop a pronounced osteopenia, although the particular drivers of this are unknown (Green et al. 2015). Outside studies suggest this is largely due to the significant decrease of IGF-1 expression, which is vital for bone growth and development (Xiao et al. 2016). FDA approval of L-glutamine (GLN) is said to reduce the acute complications of SCD by increasing antioxidant capacity of RBCs, however opposing research has led to uncertainty about the proposed therapeutic mechanism (Quinn 2018). GLN is utilized ubiquitously as a precursor for antioxidant synthesis to combat oxidative stress; therefore, the benefit experienced by sickle patients may be explained by means outside of RBC usage. Under normal conditions the liver is one of the primary sources of GLN for the body. However, the liver becomes a major site of GLN consumption during disease states to maintain homeostatic activity and reduce hepatic damage from oxidative stress (Cruzat et al. 2018). We hypothesize that GLN therapy prevents sickle bone loss by increasing liver expression of IGF-1.
METHODS: Male Townes sickle mice (SS, Jackson) and littermate controls (AA) were evaluated at 4, 8, 12 and 16 wks old. (n=10/group, IACUC approved). Half the animals received GLN in drinking water (0.7% w/v) from weaning at 4 wks old until sacrifice. Changes in femoral cortical and trabecular bone were assessed by µCT. Livers were weighed, as hepatomegaly provides a clinical index for iron storage and cirrhosis in SCD. In addition, plasma concentrations of liver derived IGF-1, the antioxidant hemopexin (HPX), and gamma-glutamyl transferase (GGT) activity were measured to determine therapeutic efficacy.
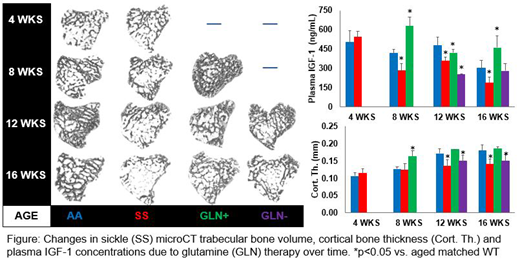
RESULTS: At 4wks there was no difference in bone, but SS livers were 20% larger than AA although liver plasma markers did not significantly differ. GLN therapy was effective at 8 wks, preventing cortical bone thinning in SS mice and reducing hepatomegaly by 12%; which remained consistent at all ages. IGF-1 plasma concentrations reflected this reduction in liver size with ~2 fold increase. HPX and GGT levels were similar to controls within treatment group, but were significantly reduced in untreated SS mice. At 12 wks, GLN effects split into two groups: (i) elevated IGF-1 (GLN+, like AA) or (ii) reduced IGF-1 (GLN-) consistent with loss of GLN effect in these mice. At 12 wks, cortical and trabecular bone in GLN+ mice group were similar to AA, while bone of GLN- mice were comparable to untreated SS mice. Protective effect of GLN on cortical bone remained by 16 wks, but was lost in trabecular bone, even in GLN+ mice. HPX concentrations followed a similar pattern, however GGT activity of GLN+ group reduced to GLN- rates.
DISCUSSION: Our studies reveal that GLN therapy increases the expression of IGF-1 and prevents the osteopenia that develops in adolescence in SCD. However, the protective effects of GLN on trabecular bone in SS mice appears to be lost with skeletal maturity. GLN therapy increases the amount of HPX available to prevent iron absorption into the tissue; however sickle RBC hemolysis still occurs during GLN therapy, thereby delaying oxidative stress derived hepatic damage as suggested by GGT at 16 wks. Why cortical and trabecular bone respond differently to GLN in SCD at maturity is not yet clear. Previous studies have shown that systemic liver derived IGF-1 preferentially affects cortical bone growth in normal mice, while locally produced IGF-1 is responsible for trabecular. In a similar manner to the liver, we speculate that iron accumulate within sickle bone may alter local bone cell expression of IGF-1.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal