

Pharmacogenomics is becoming more and more important due to personalized medicine. To understand the association between the genetic background of a patient and the response to a tailored treatment would strongly influence the choice of therapy and reduce trial-and-error strategies. Various screening assays have been established, e.g. to test the impact of acquired gene mutations in cancer on the response to different agents and combinations in cell culture models. In the era of genome data, in silico analyses are becoming an interesting tool and the hope is growing to predict a patient's response to therapy by association studies using the genetic makeup of both, the individual patient as well as the disease.
The aim of this study was to get insights into the genetic background of AML patients refractory to treatment and to investigate associations of polymorphisms (SNPs) in cancer treatment target genes and disease associated mutations contributing to an altered response to treatment.
The cohort consisted of 247 AML patients diagnosed by cytomorphology following the WHO classification. All patients were treated intensively with a standard chemotherapy protocol such as 7+3 and response to treatment was assessed after first and second induction. Following ELN guidelines patients were grouped into responder (group 1), showing cytomorphological complete remission after first induction (n=186), and treatment failure (group 2) with only partial remission or progressive disease after second induction (n=61). Whole genome sequencing (WGS) was performed with 90x coverage for all samples at diagnosis to assess their mutational profiles as well as their germline background. In depth analyses were restricted to exonic SNPs of 217 drug target genes (DrugBank 5; Schärfe et al., 2017) associated with cancer treatments and 20 recurrently mutated genes in AML. Morphologic subtypes as well as mutation frequencies in AML genes were similar in the responder and treatment failure groups. In total 9,742 unique SNPs were found with a median of 1,603 per patient. 37% of these SNPs were found in single patients only and another 27% were found in less than 5% of the cases. SNPs and somatic variants for the addressed genes were combined into a single matrix and co-occurrences of two variants were assessed by matrix multiplication for each group.
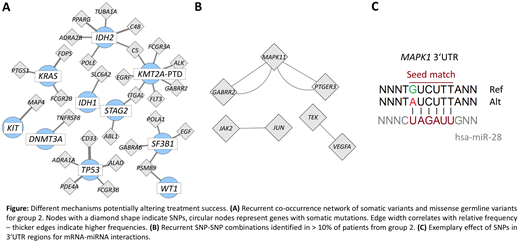
First, we were interested in group-specific co-occurrences of SNPs and somatic variants. Here we found considerably less co-occurrences in group 1 compared to group 2. All identified co-occurrences in group 1 were unique whereas 772 co-occurrences could be found in more than one patient in group 2. For group 2 a co-occurrence network was created based on recurrent combinations of somatic variants and missense germline variants (figure A). 51% of the patients from group 2 were involved in these interactions, while group 1 did not show any recurrent co-occurrences. It was interesting to note that IDH2, KMT2A-PTD, SF3B1 and TP53 recurrently co-occurred with multiple missense SNPs. Second, we addressed the association of two SNPs in the investigated drug target genes. Looking at SNPs co-occurring in at least 10% of the patients again reflected the heterogeneity of group 1, showing no recurrently co-occurring SNPs, while group 2 showed 6 SNP pairs associated with MAPK family signaling cascades (figure B). Last, it was noteworthy that multiple non-coding SNPs were found in the 3'UTR of the drug target genes. These variants showed a higher frequency in group 2 compared to group 1. SNPs in the 3'UTR region might modify mRNA - miRNA interactions by creating, abolishing, or changing the miRNA-binding sites (figure C). Recently multiple miR-SNPs have been described that were prognostic for treatment outcome, suggesting a potential as predictive biomarkers.
In conclusion, WGS data allow comprehensive genome wide but also targeted association analyses. We found three different mechanisms potentially altering treatment sensitivity based on the genetic makeup of individual patients. 1) Association of somatic mutations and SNPs located in drug target genes, 2) association of two SNPs, and 3) SNPs in the 3'UTR modulating miRNA interaction. All three mechanisms occurred only in the refractory AML cases and not in the responder group. This study shows the power of genomic data and in silico analyses that are the basis for future tools to predict response to treatment for individual patients.
Meggendorfer:MLL Munich Leukemia Laboratory: Employment. Walter:MLL Munich Leukemia Laboratory: Employment. Hutter:MLL Munich Leukemia Laboratory: Employment. Baer:MLL Munich Leukemia Laboratory: Employment. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Kern:MLL Munich Leukemia Laboratory: Employment, Equity Ownership. Haferlach:MLL Munich Leukemia Laboratory: Employment, Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal